Decoding Evolution in Real-Time: How B Cell Receptor Somatic Hypermutation Phylogenetics Powers Antibody Discovery and Disease Insights
This article provides a comprehensive analysis of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic patterns, a cornerstone of adaptive immunity and therapeutic antibody development.
Decoding Evolution in Real-Time: How B Cell Receptor Somatic Hypermutation Phylogenetics Powers Antibody Discovery and Disease Insights
Abstract
This article provides a comprehensive analysis of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic patterns, a cornerstone of adaptive immunity and therapeutic antibody development. We explore the foundational biology of SHM and affinity maturation, detailing how phylogenetic trees model the clonal evolution of B cell lineages. Methodologically, we review modern computational pipelines for reconstructing these trees from high-throughput sequencing data, emphasizing their application in tracking autoimmune, infectious, and oncological disease progression. We address common analytical challenges, such as distinguishing driver from passenger mutations and handling convergent evolution, with optimization strategies. Finally, we compare the validation frameworks and performance metrics of leading phylogenetic tools. This guide is tailored for researchers and drug developers seeking to leverage BCR phylogenetics for biomarker discovery, vaccine response assessment, and next-generation biologic design.
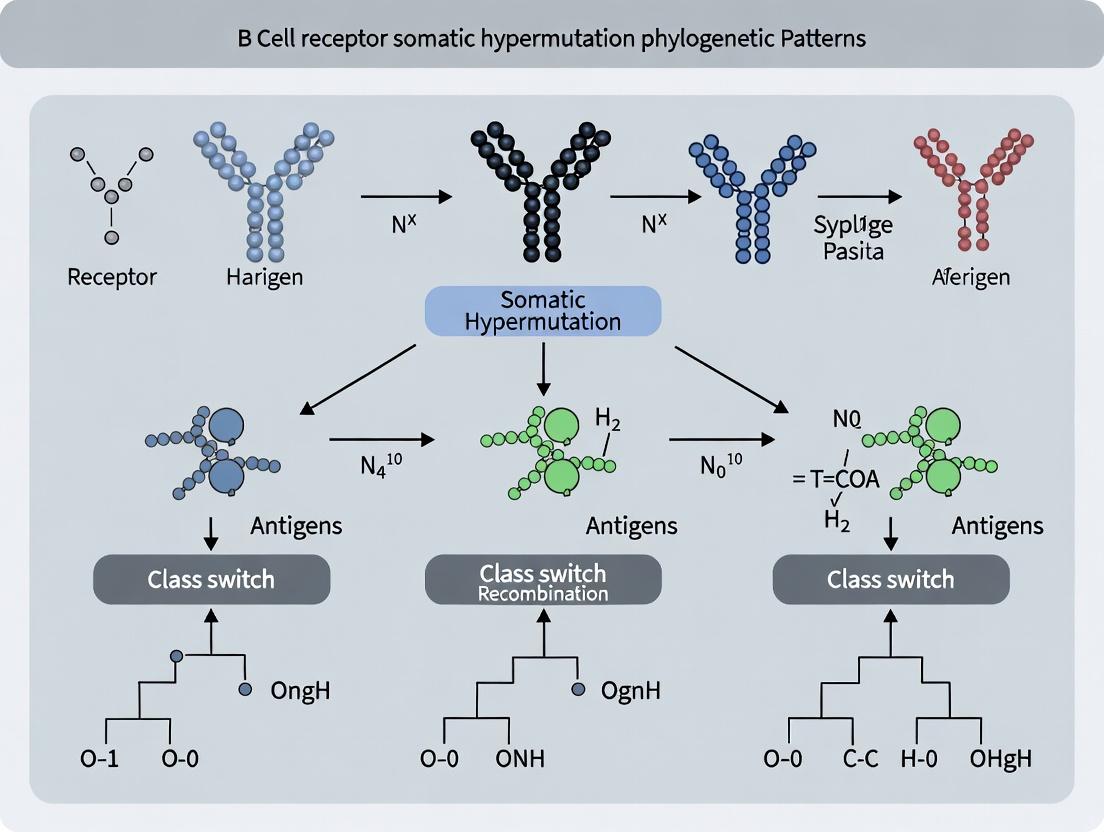
The Engine of Adaptation: Foundational Principles of B Cell Receptor Somatic Hypermutation and Phylogenetic Inference
Somatic hypermutation (SHM) and affinity maturation are the cornerstone processes by which the adaptive immune system generates high-affinity antibodies. In the context of B cell receptor (BCR) somatic hypermutation phylogenetic patterns research, understanding the precise molecular mechanisms is paramount. Phylogenetic trees reconstructed from variable gene sequences of B cell clones provide a historical record of SHM activity, allowing researchers to infer selection pressures, mutation rates, and clonal dynamics within germinal centers. This whitepaper details the current molecular model of SHM, the resultant affinity maturation, and the methodologies used to investigate them, providing a technical foundation for phylogenetic interpretation.
Core Molecular Mechanisms of Somatic Hypermutation
Initiation: AID-Mediated Deamination
The process is initiated by Activation-Induced Cytidine Deaminase (AID), which deaminates deoxycytidine (dC) to deoxyuracil (dU) within the variable region of immunoglobulin genes. This creates a U:G mismatch. AID targeting is preferential for single-stranded DNA, occurring during transcription within specific "hotspot" motifs (e.g., WRCY, where W = A/T, R = A/G, Y = C/T).
Resolution of Lesions and Mutation Outcomes
The U:G mismatch is processed by several DNA repair pathways, leading to diverse mutational outcomes:
- Replication across dU: Direct replication leads to a C→T transition mutation.
- Mismatch Repair (MMR): Recognition of the mismatch by MSH2/MSH6 recruits error-prone polymerases like Pol η, leading to mutations at adjacent A/T bases (creating "footprints").
- Base Excision Repair (BER): Uracil-N-glycosylase (UNG) removes the uracil, creating an abasic site. Subsequent error-prone synthesis by polymerases such as Pol ζ introduces mutations at the original site and nearby nucleotides.
Selection for Affinity Maturation
B cells expressing mutated BCRs compete for limited antigen presented by follicular dendritic cells and T cell help in the germinal center. Cells with higher affinity BCRs receive stronger survival signals, leading to clonal expansion and further rounds of SHM. This iterative process of mutation and selection drives affinity maturation.
Table 1: Key Quantitative Parameters of SHM in Human B Cells
| Parameter | Typical Value / Range | Notes / Context |
|---|---|---|
| Mutation Rate | ~10⁻³ to 10⁻⁴ per base per generation | ~10⁶ times higher than background genomic mutation rate. |
| Hotspot Motif Frequency | WRCY: ~4-6x higher mutation | Compared to non-hotspot sequences. |
| Transitions vs. Transversions | ~60:40 ratio | Transitions (C→T, G→A) are slightly favored. |
| A/T Mutation Frequency | ~30-40% of total mutations | Dependent on functional MMR pathway; a signature of polymerase η activity. |
| Germinal Center B Cell Division Rate | ~6-12 hours per cycle | Allows for rapid accumulation of mutations over days. |
| Typical Mutation Load in Memory B Cells | 10-30 mutations in V(D)J | Varies by antigen exposure and time; used for phylogenetic clustering. |
Table 2: Enzymes Central to SHM and Their Functions
| Enzyme / Factor | Primary Function in SHM | Consequence of Deficiency |
|---|---|---|
| AID (AICDA) | Cytidine deaminase; initiates SHM. | Complete absence of SHM and CSR. |
| UNG | Uracil-DNA glycosylase; excises dU in BER pathway. | Skewed mutations: C→T transitions dominate; loss of A/T mutations. |
| MSH2/MSH6 | Recognizes U:G mismatches; initiates MMR pathway. | Drastic reduction in A/T mutations. |
| DNA Polymerase η | Error-prone TLS polymerase; inserts mutations at A/T. | Lack of A/T mutations (as in Xeroderma Pigmentosum V variant). |
| DNA Polymerase ζ | Error-prone TLS polymerase; extends from mismatches. | Reduced mutation frequency and altered spectra. |
Experimental Protocols for SHM and Phylogenetic Analysis
Protocol: In Vitro SHM Assay Using CH12F3 Cell Line
Purpose: To quantify AID-induced mutation frequency and analyze spectra under controlled conditions.
- Culture: Maintain CH12F3 mouse B lymphoma cells in RPMI-1640 + 10% FBS.
- Stimulation: To induce AID expression, treat cells with 10 ng/mL IL-4, 10 µg/mL anti-CD40 antibody, and 1 µg/mL TGF-β for 72 hours.
- Genomic DNA Extraction: Harvest cells and extract gDNA using a silica-membrane column kit.
- Target Amplification: PCR-amplify a ~500-bp region of the Igα (Cd79a) gene, a known in vitro AID target, using high-fidelity polymerase.
- Cloning and Sequencing: Clone PCR products into a TA-vector. Transform competent E. coli. Pick 50-100 colonies for Sanger sequencing.
- Analysis: Align sequences to the germline reference. Count mutations to calculate mutation frequency (mutations/bp/division) and analyze motif context.
Protocol: Single-Cell BCR Sequencing & Phylogenetic Tree Reconstruction
Purpose: To trace the clonal lineage and SHM history of antigen-specific B cells.
- Single-Cell Sorting: Sort single, live, antigen-binding (using fluorescent antigen probes) B cells from lymphoid tissue into 96-well plates.
- Reverse Transcription & Amplification: Perform nested multiplex PCR or use template-switch-based RT-PCR to amplify full-length V(D)J heavy- and light-chain transcripts from each cell.
- High-Throughput Sequencing: Prepare libraries and sequence on an Illumina MiSeq or HiSeq platform (2x300 bp recommended).
- Bioinformatic Processing:
- Alignment & Assembly: Use tools like IgBLAST or IMGT/HighV-QUEST to assign V, D, J genes and identify CDR3 regions.
- Clonal Grouping: Cluster sequences into clonal families based on shared V/J genes and identical or highly similar CDR3 amino acid sequences.
- Phylogenetic Inference: For each clonal family, perform multiple sequence alignment of the V region. Use maximum likelihood (e.g., RAxML) or Bayesian (e.g., BEAST2) methods to infer phylogenetic trees.
- Selection Analysis: Apply models like BASELINe or dN/dS ratios to quantify positive selection in CDR vs. framework regions.
Diagrams of Signaling Pathways and Workflows
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Materials for SHM/Affinity Maturation Research
| Item | Function / Application | Example / Note |
|---|---|---|
| Recombinant AID Protein | In vitro deamination assays to study enzyme kinetics and targeting. | Human/mouse AICDA, often N-terminal His-tagged for purification. |
| AID-Deficient Mice (Aicda⁻/⁻) | In vivo control to confirm SHM-dependent phenotypes. | Foundational model for studying humoral immunity. |
| CH12F3 Cell Line | In vitro model for studying both SHM and class switch recombination (CSR). | Mouse B lymphoma line; mutation inducible by cytokine/costimulation. |
| Fluorescent Antigen Probes | Identification and sorting of antigen-specific B cells for single-cell analysis. | e.g., Recombinant HA-tagged protein + anti-HA Alexa Fluor conjugate. |
| Single-Cell BCR Amplification Kits | Robust amplification of paired heavy- and light-chain transcripts from single B cells. | Commercial kits (e.g., from Takara Bio, Bio-Rad) enhance success rate. |
| High-Fidelity DNA Polymerase | Accurate amplification of BCR genes for cloning without introducing PCR errors. | Essential for mutation frequency assays (e.g., Q5, Phusion). |
| UNG Inhibitor (UGI) | Experimental tool to dissect the BER pathway's role in SHM. | Co-expression with AID skews mutation spectrum toward C→T. |
| Error-Prone DNA Polymerase Inhibitors | Chemical tools to probe the role of specific TLS polymerases. | e.g., curcumin for Pol η inhibition (requires controlled validation). |
| Germline Gene Reference Databases | Essential bioinformatic resource for assigning mutations. | IMGT, NCBI Ig Blast. |
Within the broader thesis on B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic patterns, this guide formalizes the conceptual and technical framework for reconstructing B cell clonal expansion as an evolutionary phylogeny. The adaptive immune response is a microcosm of Darwinian evolution, where antigen-driven selection acts upon B cell clones undergoing SHM and clonal expansion. Analyzing this process through a phylogenetic lens allows researchers to trace the historical relationships between B cell variants, identify convergent evolution toward high-affinity solutions, and decode the antigenic history of a response. This is critical for understanding autoimmune diseases, vaccine efficacy, and the development of therapeutic antibodies.
Foundational Concepts and Quantitative Data
B cell phylogenies are inferred from BCR immunoglobulin heavy chain (IGH) variable region sequences. Key quantitative metrics define clonal relationships and evolutionary dynamics.
Table 1: Key Metrics for B Cell Phylogenetic Analysis
| Metric | Typical Range/Value | Interpretation |
|---|---|---|
| SHM Rate | ~10⁻³ to 10⁻⁴ mutations/base/generation | Defines the molecular clock for divergence timing. |
| Clonal Relatedness Threshold | ≥85% IGHV gene identity | Sequences within this threshold are considered potential clonal relatives. |
| Linearity Index | 0 (Perfect Tree) to 1 (Perfect Linear) | Measures tree branching structure; lower values indicate greater diversification. |
| Mean Pairwise Distance | Varies per clone (e.g., 5-30 nucleotides) | Average genetic distance between all sequences in a clonal family. |
| Selection Pressure (dN/dS) | dN/dS > 1 (Positive), ≈1 (Neutral), <1 (Negative) | Identifies antigen-driven selection in Complementarity-Determining Regions (CDRs). |
| Clonal Diversity (Shannon Index) | Clone-dependent; higher = more diverse repertoire. | Quantifies the evenness and richness of B cell clones in a sample. |
Core Methodological Pipeline
Experimental Protocol 1: Single B Cell Sorting and BCR Sequencing
- Sample Preparation: Isolate mononuclear cells (PBMCs, lymph node, or spleen tissue).
- Cell Staining: Stain with fluorescently labeled antibodies for surface markers (e.g., CD19+, CD20+, CD27+ for memory B cells) and a viability dye.
- Fluorescence-Activated Cell Sorting (FACS): Single B cells are sorted into 96- or 384-well plates containing lysis buffer.
- Reverse Transcription & PCR: Use reverse transcription with gene-specific primers for IGH constant regions, followed by nested multiplex PCR to amplify full-length IGH V(D)J rearrangements.
- Library Preparation & High-Throughput Sequencing: Add sequencing adapters and barcodes. Sequence on platforms like Illumina MiSeq/NextSeq to obtain paired-end reads.
- Bioinformatics Analysis: Process reads through pipelines like pRESTO, IgBLAST, and Change-O for annotation, error correction, and clonal grouping.
Experimental Protocol 2: Phylogenetic Tree Inference from BCR Sequences
- Clonal Family Definition: Group sequences sharing the same IGHV and IGHJ genes and having a junction region length within a 6-nucleotide tolerance.
- Multiple Sequence Alignment: Align the V(D)J region nucleotide sequences using a tool like MAFFT or Clustal Omega.
- Evolutionary Model Selection: Use jModelTest or PartitionFinder to select the best-fit nucleotide substitution model (e.g., HKY+G).
- Tree Building: Apply phylogenetic algorithms.
- Maximum Likelihood: Using RAxML or IQ-TREE (preferred for accuracy).
- Bayesian Inference: Using BEAST2 (allows for dating divergence times).
- Tree Visualization & Annotation: Use ggtree (R) or FigTree to visualize trees, annotating branches with SHM load and sequences with isotype.
Key Signaling Pathways and Workflows
Diagram 1: B Cell Activation & SHM Pathway (84 characters)
Diagram 2: BCR Phylogeny Construction Workflow (81 characters)
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for B Cell Phylogeny Studies
| Item | Function/Application |
|---|---|
| Anti-human CD19/CD20 Microbeads | Magnetic bead-based isolation of B cells from complex tissues. |
| Fluorochrome-conjugated Antibodies (CD19, CD20, CD27, CD38, IgD) | Phenotypic characterization and sorting of specific B cell subsets via FACS. |
| Single-Cell Lysis Buffer (e.g., RNase Inhibitor + DTT) | Immediate cell lysis and RNA stabilization post-sorting. |
| SMARTer Human BCR Kits | Integrated kits for cDNA synthesis and amplification of full-length IGH transcripts from single cells. |
| Illumina MiSeq Reagent Kit v3 (600-cycle) | High-throughput sequencing with read lengths sufficient for full V(D)J coverage. |
| pRESTO & Change-O Software Suites | Open-source bioinformatics pipelines for processing raw BCR-seq data, error correction, and clonal clustering. |
| IgBLAST Database | NCBI tool for annotating V, D, J gene usage and mutation analysis. |
| IQ-TREE Software | Efficient maximum likelihood phylogenetic inference with model selection. |
| ggtree R Package | Powerful tool for phylogenetic tree visualization and annotation with associated metadata. |
This whitepaper details the genetic and epigenetic machinery governing somatic hypermutation (SHM) of immunoglobulin genes in B cells, a cornerstone of adaptive immunity. Framed within broader research on B cell receptor (BCR) phylogenetic patterns, it dissects the molecular players that shape mutational landscapes, influencing antibody affinity and the evolutionary trajectories of B cell clones.
Core Mechanism: AID Initiation and Beyond
Activation-induced cytidine deaminase (AID) is the essential initiator of SHM, deaminating deoxycytidine to deoxyuidine in single-stranded DNA within the Ig variable region. This lesion seeds the mutational process, but the ultimate pattern is determined by a cascade of downstream factors.
Key Regulators and Their Functions
| Regulator | Type | Primary Function in SHM | Impact on Mutation Pattern |
|---|---|---|---|
| AICDA (AID) | Enzyme (Deaminase) | Initiates SHM by converting C to U. | Creates U:G mismatches; defines initial hotspot targeting (e.g., WRCY motifs). |
| UNG | Enzyme (Glycosylase) | Excises uracil, creating abasic sites. | Shifts mutations from C/G to Transversions at A/T bases. |
| MSH2-MSH6 (MutSα) | MMR Complex | Binds U:G mismatches, recruits translesion polymerases. | Promotes mutations at A/T pairs; expands mutational spread beyond initiation site. |
| POL η | Translesion Polymerase | Error-prone synthesis across abasic sites. | Introduces primarily A/T mutations. |
| EXO1 | Nuclease | Processes DNA ends in MMR pathway. | Facilitates error-prone patch synthesis, extending mutation footprint. |
| 14-3-3 | Adaptor Protein | Binds AID, facilitates its targeting & stabilization. | Modifies AID recruitment efficiency and potentially target specificity. |
| Spt5 | Transcription Elongation Factor | Recruits AID to transcribed genes. | Couples SHM initiation to transcription, influencing regional targeting. |
AID Targeting and the SHM Pathway
The following diagram illustrates the core SHM pathway initiated by AID and the key downstream decision points that determine mutation patterns.
Epigenetic Regulation of SHM Targeting
Epigenetic landscapes critically direct AID activity. Key regulators are summarized below.
| Epigenetic Feature | Role in SHM Targeting | Experimental Evidence |
|---|---|---|
| Histone Modifications | H3K4me3, H3K36me3, H3K79me2 correlate with SHM hotspots. | ChIP-seq shows enrichment in mutating regions. |
| DNA Methylation | Hypomethylation permits AID access; hypermethylation inhibits. | Whole-genome bisulfite sequencing of B cell subsets. |
| Chromatin Accessibility | Open chromatin (ATAC-seq peaks) at Ig loci facilitates AID binding. | ATAC-seq and AID ChIP-seq correlation. |
| Non-Coding RNA | Germline transcription produces ncRNAs that may guide AID. | RNA-seq and knockdown experiments. |
| Cohesin Complex | Loop extrusion may bring enhancers close to IgV. | Hi-C in B cells shows specific loops. |
Epigenetic Landscape Shaping SHM
This diagram outlines how epigenetic signals converge to regulate AID access and targeting.
Experimental Protocols for SHM Pattern Analysis
1In VitroSHM Assay (B Cell Culture)
Purpose: To quantify and characterize SHM patterns in activated B cells.
- B Cell Isolation: Isolate naïve human or mouse B cells from spleen/blood using negative selection magnetic beads (e.g., CD43- for mouse).
- Activation & Culture: Culture cells (1e6 cells/mL) in RPMI-1640 + 10% FBS with activation cocktail:
- Mouse: 25 µg/mL LPS + 10 ng/mL IL-4 (for 72-96 hrs).
- Human: CD40L-expressing feeder cells + 100 U/mL IL-4 + 1 µg/mL CpG ODN 2006 (for 5-7 days).
- Genomic DNA Extraction: Harvest cells. Use a column-based kit to extract high-molecular-weight genomic DNA.
- Target Amplification: Design primers flanking the IgVH CDR1-CDR2 region. Perform PCR with high-fidelity polymerase (e.g., Phusion) to minimize introduced errors.
- Sequencing & Analysis: Clone PCR products into a sequencing vector or prepare for next-generation amplicon sequencing (Illumina MiSeq). Align sequences to germline references using tools like IMGT/HighV-QUEST. Calculate mutation frequency and spectrum (RGYW/WRCY bias, A/T vs. C/G mutations).
AID Chromatin Immunoprecipitation Sequencing (ChIP-seq)
Purpose: To map genome-wide AID binding sites and correlate with epigenetic marks.
- Crosslinking & Sonication: Fix 10-50 million activated B cells (from 4.1) with 1% formaldehyde for 10 min. Quench with glycine. Lyse cells and sonicate chromatin to 200-500 bp fragments.
- Immunoprecipitation: Incubate chromatin with validated anti-AID antibody or isotype control overnight at 4°C. Capture immune complexes with Protein A/G beads.
- Library Preparation & Sequencing: Reverse crosslinks, purify DNA. Prepare sequencing library using standard kits (e.g., NEBNext). Sequence on an Illumina platform (≥ 30 million reads).
- Bioinformatics: Align reads to reference genome (e.g., mm10/hg38). Call peaks (MACS2). Co-localize with H3K4me3, H3K36me3 ChIP-seq and ATAC-seq data from same cell type.
The Scientist's Toolkit: Essential Research Reagents
| Reagent / Material | Supplier Examples | Function in SHM Research |
|---|---|---|
| Recombinant Human/Mouse AID Protein | Abcam, Sino Biological | In vitro deamination assays to study enzyme kinetics and specificity. |
| Anti-AID ChIP-grade Antibody | Cell Signaling Tech, Proteintech | Mapping genomic binding sites via ChIP-seq. |
| UNG Inhibitor (Ugi) | NEB | To block the UNG pathway in vitro or in culture, isolating C→T transition patterns. |
| MSH2-/- or UNG-/- Mouse Models | Jackson Laboratory | In vivo models to dissect the relative contribution of each repair pathway to SHM spectra. |
| B Cell Activation Cocktail | Thermo Fisher, Miltenyi Biotec | Standardized reagents (LPS, IL-4, CD40L, anti-IgM) for consistent B cell activation in vitro. |
| High-Fidelity PCR Polymerase | NEB (Phusion), Takara (PrimeSTAR) | Accurate amplification of IgV regions for sequencing without introducing polymerase errors. |
| Next-Gen Sequencing Amplicon Kit | Illumina (TruSeq), Swift Biosciences | Preparing libraries from amplified IgV regions for deep mutational profiling. |
| ATAC-seq Kit | 10x Genomics (Chromium), Illumina (Nextera) | Assessing genome-wide chromatin accessibility in primary B cell subsets. |
Integrating SHM Regulators into BCR Phylogenetics
The regulators detailed herein define the "mutational grammar" of B cell evolution. In phylogenetic analyses of BCR lineages:
- AID hotspot targeting creates predictable starting points for variation.
- UNG/MSH2 bias shapes branch lengths (mutation load) and base substitution patterns (transitions vs. transversions).
- Epigenetic heterogeneity may explain why otherwise identical germline sequences mutate at different rates in different clones.
Accurate models of B cell clonal expansion and selection must therefore account for this underlying genetic and epigenetic architecture that constrains and directs the somatic evolutionary process.
The study of B cell receptor (BCR) evolution through somatic hypermutation (SHM) is central to understanding adaptive immunity, antibody maturation, and pathogenic dysregulation in lymphomas and autoimmune diseases. Phylogenetic trees reconstructed from BCR sequences provide a quantitative historical record of clonal expansion and selection. Within the broader thesis on BCR somatic hypermutation patterns, this guide details the interpretation of three core phylogenetic features: branch lengths, topology, and signatures of selection pressure. These features, when accurately decoded, reveal the dynamics of the germinal center reaction, the efficiency of affinity maturation, and the aberrations indicative of disease.
Interpreting Phylogenetic Features
Branch Lengths: A Molecular Clock for SHM
Branch lengths in a BCR phylogeny are proportional to the number of nucleotide substitutions accumulated along that lineage. They serve as a proxy for the timing and intensity of SHM activity.
- Long branches indicate periods of rapid mutation, potentially driven by strong positive selection for affinity-enhancing mutations or, alternatively, by a relaxed selection environment allowing for the accumulation of synonymous and passenger mutations.
- Short branches suggest slower mutation rates, possibly due to stringent selection, a pause in proliferation, or differentiation into a less mutagenic state (e.g., memory or plasma cells).
- Internal vs. Terminal Branches: Long internal branches followed by rapid, bushy diversification (long terminal branches) can signify a key affinity-enhancing mutation that unlocked subsequent variant exploration.
Table 1: Interpretation of Branch Length Patterns in BCR Phylogenies
| Pattern | Biological Implication | Potential Driver |
|---|---|---|
| Uniformly long branches | Sustained, high SHM activity across the lineage | Chronic antigen exposure; Germinal center (GC) re-entry |
| Uniformly short branches | Limited SHM or recent clonal expansion | Early GC response or extrafollicular response |
| Long internal, short terminals | A key early variant dominated, limited later exploration | Strong initial selection; Clonal dominance |
| Short internal, long terminals | Rapid diversification from a recent common ancestor | Efficient GC cyclic re-entry and diversification |
| Variable terminal lengths | Heterogeneous selection pressures on different subclones | Antigen affinity differences; T cell help variability |
Topology: Mapping Clonal Expansion and Diversification
Tree topology describes the branching structure and shape, revealing the mode of clonal evolution.
- Tree Shape Metrics:
- Colless/Imbalance Index: Measures asymmetry. Highly unbalanced trees (ladder-like) suggest strong, sequential selection. Balanced, bushy trees indicate multi-forking diversification.
- Sackin Index: Measures the average path length from root to leaves. Higher values indicate more sequential evolution.
- Common Topologies:
- Linear/Chain-like: Sequential replacement of dominant variants, typical of strong, directed selection.
- Bushy/Radiating: Simultaneous exploration of many variants from a common ancestor, indicative of broad antigenic engagement or polyclonal stimulation.
- Mixed: Combines features, such as a linear trunk with bushy bursts, reflecting phases of directional selection and exploratory diversification.
Selection Pressures: Quantifying Adaptive Evolution
Selection pressure is inferred by comparing observed non-synonymous (dN) to synonymous (dS) mutation rates (dN/dS, or ω).
- ω > 1 (Positive/Diversifying Selection): Non-synonymous mutations are favored. Key signal for affinity maturation in Complementarity-Determining Regions (CDRs).
- ω ≈ 1 (Neutral Evolution): Mutations are tolerated without strong selective advantage/disadvantage. May occur in Framework Regions (FRs) or during periods of relaxed selection.
- ω < 1 (Negative/Purifying Selection): Non-synonymous mutations are removed. Strong signal for structural/functional conservation, especially in FRs.
Table 2: Site-Specific Selection Analysis (FEL/SLAC/FUBAR) Outcomes
| Analysis Result | Region Typically Affected | Interpretation in BCR Context |
|---|---|---|
| Positive Selection Sites | CDR1, CDR2, CDR3 | Active affinity maturation; Antigen-contact residues under adaptive evolution. |
| Negative Selection Sites | Framework Regions (FR1-FR4) | Structural integrity conservation; Preservation of immunoglobulin fold. |
| Differentially Selected Branches | Specific tree lineages (e.g., a long branch) | Lineage-specific adaptive events (e.g., a key class-switch event or escape mutation). |
Experimental Protocols for BCR Phylogeny Construction
Wet-Lab Protocol: BCR Sequencing from B Cell Populations
Objective: Generate high-fidelity, full-length BCR (IgH) sequences from sorted B cell subsets for phylogenetic analysis.
- Cell Sorting: Isolate single B cells or bulk populations (e.g., GC B cells, memory B cells, plasmablasts) via FACS using markers (e.g., CD19+, CD38+, CD27±).
- Nucleic Acid Extraction: Use a single-cell or bulk RNA/DNA extraction kit with RNase inhibitors.
- Reverse Transcription & PCR:
- For RNA: Perform RT-PCR using primers targeting the IgH constant region (e.g., Cγ, Cα, Cμ) or switch regions.
- For DNA (Genomic): Use multiplex PCR with V-gene family-specific forward primers and J-gene reverse primers.
- Nested PCR (Optional): To increase specificity and yield for single cells, perform a second round of PCR with internal primers.
- Library Preparation & Sequencing: Purify amplicons, fragment, and prepare libraries for long-read (PacBio, Nanopore) or high-depth short-read (Illumina) sequencing. Long-read is preferred for full-length VDJ without assembly.
- Controls: Include a clonal cell line with known BCR sequence as a positive control and no-template wells as negative controls.
Computational Protocol: Phylogenetic Tree Construction & Analysis
Objective: From raw reads to a quantified phylogenetic tree.
- Pre-processing & Alignment:
- Demultiplex reads. For short reads, use tools like
pRESTOorMiXCRfor quality filtering, merging (if paired-end), and V(D)J assignment. - Align sequences to IMGT reference V, D, J genes using
IgBLASTorChange-O.
- Demultiplex reads. For short reads, use tools like
- Clonal Lineage Definition:
- Group sequences into clonal lineages based on shared V/J genes and highly similar CDR3 nucleotide sequences (≥85% identity). Use
Change-O'sDefineClones.py.
- Group sequences into clonal lineages based on shared V/J genes and highly similar CDR3 nucleotide sequences (≥85% identity). Use
- Multiple Sequence Alignment (MSA):
- Perform a nucleotide alignment of sequences within a defined clone. Use
MAFFTorClustal Omega. Mask non-informative constant regions.
- Perform a nucleotide alignment of sequences within a defined clone. Use
- Phylogenetic Inference:
- Model Selection: Use
ModelTest-NGorjModelTest2to determine the best-fit nucleotide substitution model (e.g., HKY, GTR+Γ). - Tree Building:
- Maximum Likelihood (ML): Use
IQ-TREEorRAxMLfor robustness. Command:iqtree -s alignment.fa -m HKY+G -bb 1000 -alrt 1000. - Bayesian Inference: Use
BEAST2for incorporating a molecular clock and estimating divergence times.
- Maximum Likelihood (ML): Use
- Model Selection: Use
- Selection Pressure Analysis:
- Use the
HyPhysuite (accessed via Datamonkey web server or standalone).- FEL (Fixed Effects Likelihood): Identifies sites under pervasive positive/negative selection.
- MEME (Mixed Effects Model of Evolution): Detects episodes of intermittent positive selection.
- BUSTED (Branch-Site Unrestricted Statistical Test for Episodic Diversification): Tests for gene-wide episodic diversifying selection on at least one branch.
- Use the
- Visualization & Metrics:
- Visualize trees with
FigTree,ggtree(R), orETE3(Python). - Calculate tree shape statistics (Colless, Sackin) using the
apTreeshapeR package.
- Visualize trees with
Visualization of Workflows and Pathways
Title: BCR Phylogenetics Analysis Workflow
Title: SHM and Selection in Germinal Center
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for BCR Phylogenetic Studies
| Item / Reagent | Provider Examples | Function in BCR Phylogenetics |
|---|---|---|
| Fluorescently-Labeled Antibodies (Human/Mouse) | BioLegend, BD Biosciences, Thermo Fisher | FACS sorting of specific B cell subsets (e.g., GC, memory, naive) for clone-specific analysis. |
| Single-Cell RNA-Seq Kits (5' with V(D)J) | 10x Genomics (Chromium), BD Rhapsody | High-throughput pairing of BCR sequence with full transcriptional profile from single cells. |
| Smart-seq2/3 Reagents | Takara Bio, Illumina | For full-length, high-quality BCR sequencing from low-input or single B cells. |
| IgBLAST / IMGT Databases | NCBI, IMGT | Reference databases for accurate V(D)J gene assignment and isotype calling. |
| Phylogenetic Software (IQ-TREE, BEAST2) | Open Source | Statistical inference of maximum likelihood and Bayesian phylogenetic trees from BCR alignments. |
| HyPhy Software Suite | Datamonkey Server | Suite of tools (FEL, MEME, BUSTED) for detecting selection pressures on BCR sequences. |
| Long-Read Sequencing Kits | PacBio (SMRTbell), Oxford Nanopore | Generation of full-length, phased BCR sequences without assembly, critical for accurate phylogenies. |
| B Cell Lineage Conjugates | Tracking of B cell fate and division history in in vitro or in vivo models. |
Bridging Germinal Center Dynamics with Reconstructed Lineage Histories
This whitepaper presents a technical guide for integrating dynamic cellular processes within germinal centers (GCs) with phylogenetic lineage histories reconstructed from B cell receptor (BCR) sequences. This integration is central to a broader thesis on deciphering BCR somatic hypermutation (SHM) patterns, providing a mechanistic understanding of affinity maturation—a process critical for vaccine design, therapeutic antibody discovery, and understanding autoimmune and lymphomagenic pathologies.
Core Conceptual Framework
The adaptive immune response relies on GCs, transient microanatomical structures where B cells undergo rapid proliferation, SHM, and selection. The historical record of these events is encoded in the mutational patterns of BCR immunoglobulin genes. Reconstructing lineage trees from these sequences provides a retrospective map of clonal expansion and divergence. Bridging this static lineage history with the dynamic, spatial, and competitive events within the GC is a major computational and experimental challenge. This bridge allows researchers to infer selection pressures, cellular migration patterns, and the temporal order of key molecular events.
Key Experimental Methodologies
High-Throughput BCR Sequencing & Lineage Reconstruction
Objective: To generate accurate BCR sequence data from GC B cell subsets and reconstruct phylogenetic lineage trees. Protocol:
- Cell Sorting: Isolate single B cells from GC light zone (LZ; CD83+/GL7+/CD38lo) and dark zone (DZ; CD83-/GL7+/CD38lo) populations via fluorescence-activated cell sorting (FACS).
- Single-Cell RNA-Seq & BCR Amplification: Use commercially available single-cell platforms (e.g., 10x Genomics Chromium) for simultaneous transcriptome profiling and V(D)J sequencing. Alternatively, perform nested multiplex PCR for IgH variable regions from sorted populations.
- Bioinformatic Processing:
- Process raw sequences with tools like
pRESTOandChange-Ofor annotation, error correction, and clonal clustering. - Align V(D)J sequences using IMGT/HighV-QUEST.
- For each clonal family, perform multiple sequence alignment (e.g., with MUSCLE).
- Lineage Tree Reconstruction: Use maximum likelihood (PhyML, IgPhyML) or Bayesian (BEAST2) methods to infer phylogenetic trees. Parsimony-based tools (e.g., dnapars) are also used for efficiency.
- Annotate Trees: Map SHM patterns, isotype, and spatial origin (LZ/DZ) metadata onto tree nodes.
- Process raw sequences with tools like
Spatial Transcriptomics and Multiplexed Ion Beam Imaging (MIBI)
Objective: To correlate lineage relationships with spatial location and signaling microenvironment within intact GCs. Protocol:
- Tissue Preparation: Generate tissue sections from frozen or fixed lymph node/spleen samples.
- Spatial Transcriptomics: Use Visium Spatial Gene Expression platform to capture transcriptome-wide data from defined spatial spots (55µm diameter). Probe for genes marking GC zones (e.g., CXCR4 for DZ, CCR6 for LZ), Tfh cells (PDCD1, CXCR5), and FDCs (CR2).
- Multiplexed Protein Imaging: Perform MIBI-TOF or CODEX using antibody panels conjugated to rare-earth metals or oligonucleotides.
- Data Integration: Align spatial maps with lineage data by:
- Microdissecting regions from spatial slides for subsequent BCR-seq.
- Using computational deconvolution to infer the likely spatial origin of sequenced clones based on transcriptional signatures.
In Vivo Lineage Tracing with Barcoded B Cells
Objective: To directly track the fate and diversification of individual B cell clones over time within a GC. Protocol:
- Barcode Library Generation: Create a lentiviral library containing a diverse set of random DNA barcodes (e.g., 16-20bp).
- Adoptive Transfer: Transduce a polyclonal population of naïve B cells with the barcode library. Adoptively transfer these cells into a congenic recipient mouse.
- Immunization: Challenge the recipient with a model antigen (e.g., NP-KLH).
- Longitudinal Sampling: At multiple time points (days 7, 14, 21) post-immunization, harvest GCs and sort GC B cells and plasmablasts.
- Sequencing & Analysis: Amplify and sequence the genomic barcode alongside the BCR. This creates a direct, unambiguous link between all descendant cells and their founder.
Quantitative Data Synthesis
Table 1: Key Metrics from Integrated GC Dynamics & Lineage Studies
| Metric | Typical Value/Description | Experimental Method | Significance for Bridging Dynamics & History |
|---|---|---|---|
| SHM Rate | ~10⁻³ per base per cell division | Bulk NGS of GC B cells | Provides a molecular clock for dating divergence events in lineage trees. |
| Clonal Diversity | 10-100+ unique clones per GC | Single-cell BCR-seq | Informs on the initial seeding and ongoing competition within the GC. |
| Lineage Tree Asymmetry | High variability in branch lengths | Phylogenetic reconstruction (IgPhyML) | Indicates heterogeneous selection pressures; long branches may correlate with DZ residence. |
| Temporal Branching | Major branching events early (day 5-7) post-immunization | In vivo barcoding + longitudinal sampling | Links tree topology to specific phases of the GC reaction. |
| Spatial Zoning Correlation | DZ-enriched clones show higher SHM burden | FACS + BCR-seq or spatial transcriptomics | Directly bridges cellular location (dynamics) with mutational history. |
| Selection Strength (dN/dS) | >1 for complementarity-determining regions (CDRs) | Codon-based models on lineage trees | Quantifies antigen-driven positive selection from historical sequences. |
Table 2: Research Reagent Solutions Toolkit
| Item | Function & Application |
|---|---|
| Fluorescently-Labeled Antigens | (e.g., NP-PE, NP-APC) Used in FACS to isolate antigen-binding GC B cells based on affinity. |
| Recombinant Cytokines & Proteins | (e.g., IL-4, IL-21, CD40L) For in vitro culture systems to mimic Tfh help and study SHM/selection. |
| Photoactivatable/Photoconvertible Reporter Mice | (e.g., Kaede, Confetti B cell mice) For intravital lineage tracing and spatial fate mapping within GCs. |
| AID-CreERᵀ² x Reporter Mice | Inducible genetic labeling of cells that have undergone SHM, enabling isolation and tracking of GC-experienced lineages. |
| Biotinylated Antigens & Streptavidin Tetramers | High-affinity probes for identifying rare antigen-specific B cells pre- and post-immunization. |
| Dual-Indexed Barcoding Primers | For high-throughput, multiplexed amplification of BCR sequences from single cells or bulk populations with minimal index hopping. |
| Antibody Panels for CyTOF/MIBI | Metal-conjugated antibodies for >40-parameter protein imaging of GC architecture and cell states. |
Critical Signaling Pathways in Germinal Center Dynamics
Integrated Experimental-Analytical Workflow
The bridge between GC dynamics and reconstructed lineage histories is built on converging lines of evidence from time-resolved sequencing, spatial mapping, and direct lineage tracing. This integrated approach transforms static BCR sequence snapshots into a movie of the adaptive immune response, revealing the rules of engagement between B cells, antigen, and T follicular helpers. For drug development, this framework enables the rational design of vaccines that steer lineages toward broad neutralization and the identification of pathogenic clones in autoimmunity and lymphoma with unprecedented precision. The continued development of in vivo reporters, higher-plex spatial tools, and sophisticated phylogenetic models that incorporate selection and spatial constraints will further solidify this critical bridge.
From Sequence to Insight: Computational Pipelines and Cutting-Edge Applications in BCR Phylogenetic Analysis
This whitepaper details the essential technical pipeline for analyzing B cell receptor (BCR) repertoire sequencing data, framed within the core thesis that phylogenetic patterns derived from somatic hypermutation (SHM) are critical for understanding B cell lineage fate, antigen-driven selection, and therapeutic antibody development. The transition from raw sequencing reads to inferred phylogenetic trees encapsulates the clonal evolution and affinity maturation history of B cells, providing insights into immune responses in infection, autoimmunity, and vaccination.
The modern computational workflow consists of four interdependent stages. The quantitative outputs and key decisions at each stage are summarized below.
Table 1: Core Stages of BCR Repertoire Phylogenetic Analysis
| Stage | Primary Input | Key Outputs & Metrics | Common Tools (2024) | Impact on Downstream Phylogeny |
|---|---|---|---|---|
| 1. Pre-processing & Annotation | Raw FASTQ files (IgG/IgA/IgM) | Filtered reads, V(D)J gene calls, CDR3 amino acid sequence. | MiXCR, IMGT/HighV-QUEST, pRESTO | Defines fundamental sequence identity; errors propagate. |
| 2. Clonal Grouping | Animated sequences (from Stage 1) | Clonal families (clonotypes), defined by shared V/J genes and CDR3 similarity. | Change-O, scRepertoire, partis | Determines which sequences are compared phylogenetically. |
| 3. SHM Analysis & Lineage Refinement | Sequences per clonal family | Mutation frequency, isotype, evidence of selection (dN/dS > 1). | IgPhyML, dNdScSeq, Alakazam | Identifies signals of antigen-driven selection within lineages. |
| 4. Phylogenetic Tree Reconstruction | Aligned SHM-containing sequences per lineage | Rooted phylogenetic trees, internal node sequences. | IgPhyML, RAxML-NG, FastTree | Visualizes lineage relationships and infers ancestral BCR states. |
Table 2: Quantitative Benchmarks for Clonal Grouping (Recent Studies)
| Grouping Method | Typical CDR3 Nucleotide Identity Threshold | Key Consideration | Reported Clonal Family Size Range |
|---|---|---|---|
| Single-linkage clustering | 85-90% | Sensitive to sequencing errors; requires prior error correction. | 2 - 500+ sequences |
| Hierarchical clustering | Adaptive (e.g., 90-97%) | Can better handle intra-clonal diversity from SHM. | 2 - 200+ sequences |
| Network-based | N/A (uses graph) | Effective for highly mutated repertoires (e.g., chronic infection). | Highly variable |
Detailed Experimental Protocols
Protocol 3.1: Core V(D)J Annotation Pipeline Using MiXCR Objective: To align bulk or single-cell BCR sequencing reads to germline V, D, and J gene segments and extract CDR3 regions.
- Data Input: Paired-end FASTQ files (R1, R2). Quality check with FastQC.
- Alignment & Assembly: Execute:
mixcr analyze shotgun --species hs --starting-material rna --contig-assembly --only-productive [sample_R1.fastq] [sample_R2.fastq] [output_prefix]. - Export Clonotypes: Generate a clonotype table:
mixcr exportClones --chains IGH --preset full [output_prefix.clns] [output_prefix.clones.txt]. This file contains counts, fractions, V(D)J assignments, and CDR3 sequences. - Quality Filtering: Filter the table to include only productive, high-confidence sequences (e.g., removing sequences with STOP codons in CDR3).
Protocol 3.2: Clonal Grouping with Change-O/Immcantation Suite Objective: To group annotated sequences into clonal families based on shared V/J genes and homologous CDR3 regions.
- Input Preparation: Format the annotation output into a Change-O compliant tab-separated file.
- Define Clones: Run the
DefineClones.pyscript with a distance threshold:DefineClones.py -d [data_file] --act set --model ham --norm len --dist 0.10. This uses a 90% identity threshold (dist=0.10) on the normalized Hamming distance of CDR3 nucleotides. - Output: A new column (
CLONE) is added to the file, assigning a unique identifier to each inferred clonal family.
Protocol 3.3: Phylogenetic Reconstruction with IgPhyML Objective: To infer a maximum-likelihood phylogenetic tree from a family of SHM-containing BCR sequences, using a specialized substitution model for Ig sequences.
- Alignment per Clone: For each large clonal family, create a multiple sequence alignment of the V(D)J region (focus on the V gene from FR1 through FR3). Use MUSCLE or MAFFT.
- Model Selection: Prepare a control file for IgPhyML specifying the
IGHlocus and theM0(global dN/dS) orMG(gene-specific dN/dS) evolutionary model. - Tree Inference: Execute IgPhyML:
igphyml -i [alignment.fasta] -m M0 --run_id [clone_id]. - Output: Newick format tree file (
[alignment.fasta]_phyml_tree.txt) and a stats file with branch lengths, support values, and dN/dS estimates.
Visualization of Workflows and Relationships
Diagram 1: Core BCR Phylogenetic Analysis Pipeline
Diagram 2: SHM & Selection Analysis Logic Flow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents & Tools for Experimental BCR Sequencing
| Item Name | Provider/Example | Primary Function in BCR Workflow |
|---|---|---|
| 5' RACE or V(D)J-specific Primer Panels | SMARTer Human BCR Kit (Takara Bio), NEBNext Immune Seq Kit (NEB) | Amplifies the highly variable V(D)J region from cDNA for Illumina library prep, ensuring full-length coverage. |
| Unique Molecular Identifiers (UMIs) | Integrated into commercial kits (e.g., 10x Genomics) | Short random nucleotide tags added during reverse transcription to correct for PCR amplification bias and errors. |
| Single-Cell Barcoding Reagents | 10x Genomics Chromium Controller & 5' v2 Kit, BD Rhapsody | Enables high-throughput pairing of heavy and light chains from individual B cells, crucial for monoclonal antibody discovery. |
| Spike-in Control Cells | Cell Ranger Immune Profiling Demon (10x Genomics) | Provides a known reference for assessing library complexity, sequencing sensitivity, and assay performance. |
| Human/Mouse Ig Isotype-specific Panels | BioLegend Isotyping Panels, SouthernBiotech Antibodies | Used in flow cytometry or CITE-seq to sort or tag B cells by isotype (IgG, IgA, etc.) prior to sequencing. |
| Benchmarking Synthetic BCR Libraries | ARCTIC (Synthetic Immune System) Consortium Standards | Known, designed BCR sequences used as spike-ins to validate and calibrate bioinformatics pipelines for accuracy. |
Within the burgeoning field of B cell receptor (BCR) repertoire analysis, understanding the phylogenetic patterns imprinted by somatic hypermutation (SHM) is paramount. This whitepaper provides an in-depth technical guide to four leading software toolkits—IgPhyML, dnaml, Partis, and SCOPER—that are critical for reconstructing and analyzing BCR evolutionary histories. Their application is central to a broader thesis investigating how SHM-driven phylogenies reveal trajectories of affinity maturation, clonal selection, and their implications for vaccine design and therapeutic antibody development.
IgPhyML
A specialized extension of the phylogenetic framework PhyML, IgPhyML incorporates models of SHM biology. It employs codon substitution models that account for the enzyme-driven, context-dependent nature of mutations introduced by activation-induced cytidine deaminase (AID), providing a more accurate reconstruction of BCR lineage trees.
dnaml (from PHYLIP)
A foundational maximum likelihood program for DNA sequence evolution. In BCR analysis, it is often used with standard nucleotide substitution models. While not BCR-specific, it serves as a benchmark or baseline for phylogenetic inference when simpler evolutionary models are appropriate.
Partis
A comprehensive toolkit for BCR repertoire analysis. Its core functionality includes V(D)J annotation, clonal clustering, and lineage tree inference. Partis uses a hidden Markov model (HMM)-based method for annotation and a sophisticated probabilistic framework for clustering and phylogenetics that integrates SHM information.
SCOPER
A computational method specifically designed for identifying Somatic Clones Of PERsisting B cells from bulk BCR repertoire sequencing data. It focuses on accurately clustering sequences into clonal families, a prerequisite for any downstream phylogenetic analysis.
Quantitative Data Comparison
Table 1: Core Software Features & Requirements
| Feature | IgPhyML | dnaml (PHYLIP) | Partis | SCOPER |
|---|---|---|---|---|
| Primary Purpose | BCR-specific phylogenetics | General DNA phylogenetics | BCR annotation, clustering, phylogeny | Clonal clustering (persistent cells) |
| Key Algorithm | Codon-based ML with SHM models | Nucleotide-based ML | HMM annotation, probabilistic clustering | K-means++/DBSCAN on CDR3 features |
| SHM-Aware | Yes (explicitly models) | No (standard models) | Yes (implicitly in models) | Indirectly (via clustering) |
| Input | Aligned codon sequences | Aligned DNA sequences | Raw FASTQ/FASTA reads | Annotated sequence tables (CSV) |
| Output | Phylogenetic tree, likelihood scores | Phylogenetic tree | Clusters, annotated sequences, trees | Clonal clusters, persistence calls |
| Typical Runtime | Moderate-High | Low-Moderate | High (full pipeline) | Low-Moderate |
Table 2: Application in a Standard SHM Phylogenetic Workflow
| Analysis Stage | Recommended Tool(s) | Key Metric | Expected Output for Thesis Research |
|---|---|---|---|
| Raw Data Processing & Annotation | Partis | Annotation accuracy (%) | Correct V/D/J gene assignments per read. |
| Clonal Family Clustering | Partis, SCOPER | Cluster purity, recall | Sets of sequences descended from a common naive B cell. |
| Multiple Sequence Alignment | MAFFT (used with IgPhyML/dnaml) | Alignment score | Nucleotide/codon alignment for tree building. |
| Phylogenetic Tree Inference | IgPhyML (primary), dnaml (baseline) | Tree likelihood, SHM pattern fit | Lineage trees depicting SHM pathways. |
| Tree Analysis & Visualization | FigTree, custom scripts | Tree shape statistics, branch lengths | Quantification of convergence, selection pressure. |
Experimental Protocols for BCR SHM Phylogenetic Analysis
Protocol 1: End-to-End Lineage Reconstruction with Partis and IgPhyML
This protocol details the process from raw sequencing data to a refined phylogenetic tree.
- Data Preparation: Obtain paired-end Illumina FASTQ files from sorted B cell populations.
- Annotation & Clustering with Partis:
- Command:
partis annotate --infile input.fasta --outfile annotated.csv - Command:
partis partition --infile annotated.csv --outfile clusters.yaml - This step identifies clonal families based on shared V/J genes and CDR3 homology.
- Command:
- Alignment Generation: Extract nucleotide sequences for a target clonal cluster. Perform multiple sequence alignment using MAFFT:
mafft --auto cluster_seqs.fasta > cluster_aligned.fasta. - Phylogenetic Inference with IgPhyML:
- Convert alignment to PHYLIP format.
- Run IgPhyML with a context-dependent model (e.g., GY94 with kappa/λ context parameters):
igphyml -i cluster_aligned.phy -m GY -t 3 -c kappa.
- Tree Validation: Assess tree confidence via bootstrap analysis (e.g., 100 replicates) within IgPhyML.
Protocol 2: Benchmarking Clonal Clustering with SCOPER
This protocol assesses the performance of clonal grouping, a critical step affecting downstream tree accuracy.
- Input Preparation: Generate a truth set of known clonal families from simulated BCR data or well-characterized spike-in controls.
- Run SCOPER: Execute SCOPER on the annotated sequence data using default or optimized parameters for CDR3 amino acid and nucleotide distance thresholds.
- Command (example):
scoper cluster --data input.csv --mode dbscan --output clusters.json
- Command (example):
- Performance Calculation: Compare SCOPER's clusters to the truth set. Calculate precision (true positives / predicted cluster size) and recall (true positives / actual cluster size) for each clonal family.
- Comparison: Run Partis partitioning on the same dataset and compute identical metrics to enable direct comparison of clustering fidelity.
Visualizations
Diagram 1: BCR SHM Phylogeny Analysis Workflow
Diagram 2: SHM-Aware vs. Standard Nucleotide Model in Tree Inference
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for BCR Repertoire Sequencing & Analysis
| Item | Function in BCR SHM Research | Example/Note |
|---|---|---|
| Sorted B Cell Populations | Source of genetic material. Enables tracking of SHM in specific subsets (e.g., memory, plasmablasts). | FACS-sorted CD19+/CD27+ memory B cells. |
| 5' RACE or Multiplex PCR Primers | Amplifies the variable region of BCR transcripts for sequencing. Bias affects clonal representation. | SMARTer Human BCR IgG H/K/L primers. |
| High-Fidelity Polymerase | Critical for accurate amplification with minimal PCR error, which can be mistaken for SHM. | Q5 Hot Start Polymerase. |
| Unique Molecular Identifiers (UMIs) | Short random nucleotide tags added to each cDNA molecule to correct for PCR amplification errors and duplicates. | 12nt UMI in sequencing adapters. |
| Spike-in Control Libraries | Synthetic BCR sequences with known mutations/clonal relationships. Essential for benchmarking tool accuracy (clustering, tree inference). | Custom-designed clonal lineages. |
| Reference Germline Database | Comprehensive set of V, D, J gene alleles. Required for accurate annotation of unmutated precursors. | IMGT database, partis-built germline sets. |
| High-Performance Computing (HPC) Cluster | Partis, IgPhyML, and large-scale analyses are computationally intensive, requiring significant RAM and CPU hours. | 64+ GB RAM, 16+ cores per job. |
This whitepaper provides a technical guide for applying phylogenetic methods to the study of B cell receptor (BCR) somatic hypermutation (SHM) patterns in response to persistent viral infections, specifically SARS-CoV-2 and HIV. The analysis is framed within the broader thesis that phylogenetic reconstruction of BCR lineages reveals fundamental principles of affinity maturation, convergent antibody solutions, and escape mutant evolution, with direct implications for vaccine and therapeutic antibody design.
Core Phylogenetic Concepts in B Cell Evolution
BCR evolution within germinal centers is a Darwinian process driven by SHM and selection. Phylogenetic trees reconstructed from longitudinally sampled BCR sequences map the historical relationships between B cell clones, identifying key mutations, evolutionary rates, and selection pressures.
Quantitative Comparison of SARS-CoV-2 and HIV Antibody Phylogenetics
Table 1: Key Phylogenetic Metrics for SARS-CoV-2 vs. HIV Antibody Lineages
| Metric | SARS-CoV-2 Neutralizing Antibodies (e.g., Anti-RBD) | HIV Broadly Neutralizing Antibodies (e.g., VRC01-class) | Analytical Implication |
|---|---|---|---|
| SHM Rate (per seq, per year) | 0.5 - 1.5 x 10⁻³ | 5 - 15 x 10⁻³ | HIV requires more extensive maturation. |
| Tree Depth (Avg. branch length) | Moderate (0.02-0.08 subs/site) | High (0.08-0.20 subs/site) | Indicates duration/intensity of selective pressure. |
| Convergent Solutions | High frequency in public clonotypes. | Lower frequency, require rare SHM pathways. | Vaccine design feasibility. |
| Selection Pressure (dN/dS ratio) | Strong positive in CDRs (2.5-4.0). | Very strong positive in CDRs (3.0-6.0). | Identifies functionally critical residues. |
| Lineage Latency Period | Weeks to months post-infection/vaccination. | Years post-infection. | Informs sampling strategy for lineage isolation. |
Detailed Methodological Protocols
Protocol: Longitudinal BCR Repertoire Sequencing & Lineage Assembly
Objective: To reconstruct the phylogenetic history of antigen-specific B cell lineages.
- Sample Collection: Isolate PBMCs or tissue (e.g., lymph node, bone marrow) at multiple time points post-infection/vaccination (HIV: years; SARS-CoV-2: months).
- Antigen-Specific B Cell Sorting: Use fluorescently labeled recombinant antigen (e.g., SARS-CoV-2 Spike trimer, HIV Env gp140) to sort single antigen-binding memory B cells or plasmablasts via FACS.
- Single-Cell RNA Sequencing & BCR Amplification: Use kits (e.g., 10x Genomics 5' Immune Profiling) for V(D)J enrichment or perform nested RT-PCR with V-gene and C-gene primers.
- Bioinformatic Processing:
- Processing: Use Cell Ranger (10x) or pRESTO for read quality control, assembly, and annotation of heavy and light chain sequences.
- Lineage Clustering: Group sequences into clonal lineages using hierarchical clustering based on V/J gene identity and CDR3 nucleotide similarity (≥85%).
- Multiple Sequence Alignment: Perform codon-aware alignment (MAFFT, Clustal Omega) for each lineage.
- Phylogenetic Reconstruction: Build maximum-likelihood trees (IQ-TREE, RAxML) using the GTR+Γ substitution model. Root trees using the inferred germline sequence (IgBLAST against IMGT).
- Selection Analysis: Apply PAML (CodeML) or HyPhy (FEL, MEME) to aligned lineage sequences to calculate dN/dS ratios and identify sites under positive selection.
Protocol:In VitroAffinity Maturation Replay
Objective: To experimentally validate inferred phylogenetic pathways.
- Ancestral Node Gene Synthesis: Synthesize genes encoding putative ancestral antibodies at key nodes of the phylogenetic tree.
- Yeast or Phage Display Library Construction: Introduce diversity around the ancestral sequence using error-prone PCR or oligonucleotide-directed mutagenesis targeting regions identified under positive selection.
- Selection Pressure: Perform sequential rounds of panning against the antigen under increasing stringency (e.g., decreasing antigen concentration, adding soluble competitor).
- Pathway Analysis: Sequence output pools after each round. Construct a phylogenetic tree from all output sequences to visualize the in vitro evolutionary trajectories and compare with in vivo trees.
Visualizations
Title: BCR Phylogenetics Experimental Workflow
Title: Germinal Center SHM and Selection Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for BCR Phylogenetics Studies
| Item | Function/Application | Example/Supplier |
|---|---|---|
| Recombinant Antigen (Biotinylated) | Fluorescent labeling for FACS sorting of antigen-specific B cells. | SARS-CoV-2 Spike S2P trimer; HIV BG505 SOSIP.gg. |
| Single-Cell BCR Amplification Kit | Amplification of paired heavy and light chain V(D)J from single B cells. | 10x Genomics Chromium Next GEM 5'; Takara SMARTer Human BCR. |
| High-Fidelity Polymerase | Error-free amplification for cloning ancestral antibody genes. | Q5 (NEB), KAPA HiFi. |
| Yeast Display System | In vitro affinity maturation and functional screening. | pYD1 vector; Turbo酵母 library kit. |
| Bioinformatics Pipeline | Processing raw sequences to phylogenetic trees. | Immcantation (pRESTO, Change-O, IgPhyML); PHYLIP. |
| Codon-Optimized Gene Fragments | Synthesis of inferred ancestral antibody sequences for testing. | IDT gBlocks, Twist Biosynthesis. |
| HEK293F Cells | Transient transfection for high-yield antibody production. | Thermo Fisher Expi293F System. |
| BLI/SPR Instrument | Quantifying binding kinetics (Kon, Koff, KD) of lineage members. | Sartorius Octet; Cytiva Biacore. |
This whitepaper provides a technical guide within the broader thesis of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic pattern research. The clonal expansion and somatic evolution of B cells are central to both effective immunity and pathogenesis. In autoimmunity, self-reactive clones evade normal checkpoints, while in B cell malignancies, oncogenic events drive clonal proliferation. The precise identification and characterization of these pathogenic clones through their BCR repertoire and mutation phylogenies is critical for diagnostic, prognostic, and therapeutic development.
Core Concepts: BCR Phylogenetics and Clonal Dysregulation
B cell clones originate from a common progenitor. Upon antigen exposure, clones undergo affinity maturation in germinal centers, a process driven by SHM and clonal selection. Phylogenetic trees reconstructed from BCR sequences map this evolutionary history.
- Autoimmunity: Pathogenic clones often show signs of antigen-driven selection, with shared ("stereotyped") BCRs across patients, elevated SHM, and phylogenetic patterns indicating chronic activation.
- B Cell Cancers: Malignant clones are identified by a dominant, unique BCR sequence (clonal V(D)J rearrangement) that constitutes a high fraction of the repertoire. Subclonal heterogeneity, revealed by phylogenetic branching, indicates tumor evolution and therapy resistance.
Quantitative Landscape of Pathogenic Clones
Table 1: Key Quantitative Metrics for Pathogenic Clone Identification
| Metric | Autoimmunity (e.g., SLE, RA) | B Cell Cancers (e.g., CLL, DLBCL) | Measurement Technique |
|---|---|---|---|
| Clonal Frequency | Moderate (0.1% - 5% of repertoire) | Very High (Often >20% of repertoire) | High-throughput Sequencing (HTS), Flow Cytometry |
| SHM Burden | High (5-20 mutations/V region) | Variable: CLL (Low/High), DLBCL (High) | IgBLAST, IMGT/HighV-QUEST |
| Clonality Index | Elevated (Polyclonal skew) | Highly Elevated (Monoclonal/ Oligoclonal) | Shannon Entropy, D50 Index |
| V Gene Bias | Yes (e.g., VH4-34 in SLE) | Yes (e.g., IGHV1-69 in CLL) | V/J Gene Usage Analysis |
| Intraclonal Diversity | Present (ongoing mutation) | Present in some (Subclones) | Phylogenetic Tree Analysis |
| CDR3 Characteristics | Often longer, charged | Can be stereotyped (CLL) | CDR3 Length, Amino Acid Property Analysis |
Table 2: Current Detection Method Sensitivities
| Method | Detection Limit | Primary Application | Throughput |
|---|---|---|---|
| Next-Gen Sequencing (BCR-seq) | 0.01% - 0.1% | Discovery, Minimal Residual Disease (MRD) | High |
| Flow Cytometry | 0.1% - 1% | Diagnostic screening, Phenotyping | Medium |
| ddPCR (Assay-specific) | 0.001% - 0.01% | Ultra-sensitive MRD monitoring | Low-Medium |
| Single-Cell BCR-seq | N/A (Single Cell) | Paired heavy/light chain, Phylogenic tracing | Medium |
Experimental Protocols for Clone Identification
High-Throughput BCR Repertoire Sequencing (BCR-Seq)
Objective: To comprehensively profile the BCR immunoglobulin heavy chain (IGH) repertoire from bulk tissue or sorted B cells.
Protocol:
- Sample Prep: Isolate PBMCs or tissue mononuclear cells. Extract total RNA or genomic DNA.
- Library Construction: Use multiplex PCR with V gene family-forward and J gene-reverse primers. Include unique molecular identifiers (UMIs) to correct for PCR errors and duplication.
- Sequencing: Perform paired-end sequencing (2x300bp MiSeq or NovaSeq) to ensure full CDR3 coverage.
- Bioinformatic Analysis:
- Preprocessing: Demultiplex, merge reads, and correct via UMIs.
- Alignment & Assembly: Align to IMGT reference V, D, J genes using IgBLAST or MiXCR.
- Clonal Grouping: Cluster sequences with identical V/J genes and >85% CDR3 nucleotide identity.
- Phylogenetic Analysis: For each clone, perform multiple sequence alignment (Clustal Omega) and reconstruct maximum-likelihood trees (RAxML, IgPhyML).
Single-Cell BCR and Transcriptome Sequencing
Objective: To link clonal BCR sequence with the cell's full transcriptional phenotype.
Protocol:
- Single-Cell Sorting: Use FACS to index-sort single B cells into 96- or 384-well plates or employ droplet-based partitioning (10x Genomics).
- Libraries: For plate-based: Perform nested RT-PCR for IGH and IGK/L chains. For droplet-based: Use commercially available kits (10x Genomics 5' Immune Profiling).
- Analysis: Process transcriptome data (Cell Ranger). Assemble BCR contigs (Cell Ranger VDJ). Tools like Scirpy enable integrated clonotype analysis within the transcriptomic cluster landscape.
Functional Validation of Pathogenicity
Objective: To test the autoreactivity or oncogenic potential of a identified BCR clone.
Protocol (Autoimmunity - HEp-2 IF assay):
- Cloning: Clone the identified heavy and light chain variable regions into IgG1 expression vectors.
- Recombinant Antibody Production: Co-transfect heavy and light chain plasmids into Expi293F cells. Purify antibody via Protein A/G.
- Immunofluorescence: Apply purified recombinant antibody to fixed HEp-2 cell slides. Detect with anti-human IgG-FITC. ANA-positive staining confirms self-reactivity.
Visualization of Workflows and Pathways
Title: BCR Clone ID & Phylogenetic Analysis Workflow
Title: Pathogenic B Cell Clone Survival & Expansion Signals
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Pathogenic B Cell Clone Research
| Reagent Category | Specific Item/Kit | Primary Function in Research |
|---|---|---|
| Sample Prep & Isolation | Human CD19+ B Cell Isolation Kit (Magnetic Beads) | Negative selection for pure, untouched B cell populations from PBMCs or tissue. |
| BCR Sequencing | SMARTer Human BCR IgG IgM H/K/L Profiling Kit (Takara) | Multiplex PCR for comprehensive NGS library prep from RNA with UMI integration. |
| Single-Cell Profiling | Chromium Next GEM Single Cell 5' Kit with Feature Barcode (10x Genomics) | Integrated single-cell gene expression and paired V(D)J profiling. |
| Antibody Expression | Expi293 Expression System (Thermo Fisher) | High-yield transient transfection for recombinant monoclonal antibody production. |
| Functional Assays | HEp-2 ANA Substrate Slides (Euroimmun) | Gold-standard substrate for detecting antinuclear autoreactivity of recombinant antibodies. |
| Flow Cytometry | Anti-human CD19, CD27, CD38, IgD, BCMA Antibodies | Phenotypic characterization of B cell subsets (naïve, memory, plasma blasts) and clones. |
| Bioinformatics | IMGT/HighV-QUEST, MiXCR, IgPhyML Software | Standardized analysis pipeline for annotating sequences, clustering clonotypes, and phylogenetic reconstruction. |
| Cytokines/Stimuli | Recombinant human BAFF, IL-4, IL-21, CpG ODN | In vitro stimulation to mimic survival and differentiation signals promoting pathogenic clones. |
This whitepaper details the integration of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic analysis into rational drug and vaccine design. Framed within a broader thesis on SHM phylogenetic patterns, this guide provides a technical roadmap for leveraging evolutionary insights to engineer superior monoclonal antibodies (mAbs) and predict immunogen success.
Foundational Concepts: SHM and Lineage Tracing
Somatic hypermutation in B cells, driven by Activation-Induced Cytidine Deaminase (AID), introduces point mutations into immunoglobulin variable region genes during affinity maturation. Phylogenetic reconstruction of these mutations allows for the inference of ancestral BCR states and the evolutionary trajectory toward high affinity and breadth.
Key Data from Recent Studies
Quantitative insights from recent research (2023-2024) are summarized below.
Table 1: Phylogenetic Metrics Correlated with Antibody Developability & Efficacy
| Metric | Definition | Correlation with Outcome | Typical Value Range (High-Performing Lineages) | Source (Example Study Focus) |
|---|---|---|---|---|
| Lineage Depth | Number of mutations from inferred germline ancestor to mature antibody. | Moderate positive correlation with affinity; beyond a threshold, correlates with autoreactivity risk. | 15-35 nucleotide substitutions | HIV bnAb development |
| Branching Factor | Average number of child nodes per node in lineage tree. | High branching indicates robust clonal expansion and selection, predictive of antigen immunodominance. | 1.8 - 2.5 | Influenza vaccine response |
| Convergent Mutation Rate | Frequency of identical amino acid mutations appearing independently in multiple sub-lineages. | High rate indicates strong selective pressure and identifies critical functional sites for epitope targeting. | 3-7 key convergent sites per lineage | SARS-CoV-2 RBD-targeting Abs |
| Selection Pressure (dN/dS) | Ratio of non-synonymous to synonymous mutation rates. | dN/dS > 1 in Complementarity-Determining Regions (CDRs) indicates positive selection for affinity. | CDR: 1.5-3.0; Framework: ~0.5 | Broadly neutralizing antibody (bnAb) discovery |
| Ancestor Neutralization Breadth | Percentage of viral variants neutralized by the inferred unmutated common ancestor (UCA). | High UCA breadth predicts feasible vaccine elicitation pathways. | 10-40% for complex pathogens | HIV-1 VRC01-class bnAbs |
Table 2: Impact of Phylogenetic-Informed Design on mAb Properties
| Design Strategy | Typical Improvement vs. Lead Candidate | Reduction in Development Risk | Application Example |
|---|---|---|---|
| Ancestor Maturation | 2-5x increased expression titer in CHO cells | High (improved biophysical properties) | Anti-IL-23p19 clinical candidate |
| Consensus Sequence | 10-50% increase in neutralization breadth | Moderate to High | Pan-coronavirus mAbs |
| Branch Resampling | Identifies variants with 1-2 log lower polyspecificity (PSR assay) | High (reduced attrition due to off-target binding) | CNS-targeting therapeutics |
Experimental Protocols
Protocol: Single B Cell Sequencing & Lineage Reconstruction
Objective: To obtain paired heavy- and light-chain sequences from antigen-specific B cells and reconstruct their phylogenetic lineage.
Materials: See "The Scientist's Toolkit" below. Procedure:
- Cell Sorting: Isolate single antigen-specific B cells (using fluorescently labeled antigen probes or memory B cell markers) via Fluorescence-Activated Cell Sorting (FACS) into 96-well plates containing lysis buffer.
- Reverse Transcription & PCR: Perform nested multiplex PCR using V gene-specific primers to amplify IgH and IgL chain transcripts.
- Sequencing & Annotation: Sequence amplicons via high-throughput sequencing. Annotate V(D)J genes, mutation counts, and CDR3 sequences using tools like IMGT/HighV-QUEST or partis.
- Lineage Inference: For cells from the same clonal family, align variable region sequences. Use tools like IgPhyML or Dowser to:
- Infer the Unmutated Common Ancestor (UCA).
- Build a maximum-likelihood phylogenetic tree.
- Calculate dN/dS and identify sites under positive selection.
Protocol:In SilicoAffinity Maturation Simulation
Objective: To guide antibody engineering by simulating evolutionary paths.
Procedure:
- Define Starting Sequence: Input UCA or intermediate ancestor sequence.
- Model SHM: Use a probabilistic model (e.g., from S5F mutation data) to generate a library of in silico variants, focusing mutations on CDRs.
- Affinity Prediction: Score variants using a trained neural network (e.g., DeepAb, AntiBERTy) or molecular dynamics/docking (e.g., Rosetta) for binding energy.
- Select & Iterate: Apply a selection filter (e.g., top 0.1% predicted affinity) to "surviving" variants. Use them as parents for the next round of simulated mutation. Repeat for 3-5 cycles.
- Synthesize Top Candidates: Express and test the highest-scoring in silico-evolved variants.
Visualization of Workflows and Pathways
Title: From B Cell to Phylogenetic Application Workflow
Title: BCR Signaling to AID Activation Pathway
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for Phylogenetic-Driven Discovery
| Item | Function & Application | Example Vendor/Product |
|---|---|---|
| Fluorescent Antigen Probes | For FACS sorting of antigen-specific B cells or plasmablasts. Crucial for obtaining the relevant sequences. | Recombinant antigens conjugated to PE, APC, or BV421. |
| Single-Cell RNA-seq Kits (5' V(D)J enriched) | Captures paired full-length Ig transcripts and cell's transcriptional state from single cells. | 10x Genomics Chromium Next GEM Single Cell 5', BD Rhapsody with AbSeq. |
| Ig Isotype & Subclass Detection Antibodies | To assess class switch events within a lineage, informing immunogen design. | Anti-human IgG/IgA/IgM, IgG1-4 specific antibodies. |
| Recombinant AID (Active) | For in vitro SHM assays to validate mutation hotspots or test immunogen selection. | Purified human AICDA protein. |
| HEK293F or ExpiCHO-S Cells | Mammalian expression systems for high-throughput transient expression of ancestral/engineered antibody variants. | Thermo Fisher, Gibco systems. |
| Biosensor Chips (e.g., SPR, BLI) | For high-throughput kinetic screening (kon, koff, KD) of lineage member antibodies. | Cytiva Series S CMS chips, FortéBio Streptavidin (SA) biosensors. |
| Polyreactivity/Specificity Reagents | To screen for autoreactivity risk in engineered candidates (e.g., HEp-2 cell ELISA, lipid array). | MBL HEp-2 Substrate Slides, ANA Pattern ELISA Kits. |
Navigating Analytical Challenges: Best Practices for Robust BCR Phylogenetic Reconstruction
Within the context of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic pattern research, accurately distinguishing true somatic mutations from artifacts introduced by polymerase chain reaction (PCR) amplification and next-generation sequencing (NGS) errors is paramount. Misclassification can lead to erroneous phylogenetic trees, flawed lineage tracing, and incorrect conclusions regarding clonal selection and affinity maturation. This guide outlines rigorous, multi-layered strategies to resolve this critical ambiguity, enabling high-fidelity analysis of BCR repertoires for basic immunology and therapeutic antibody discovery.
A clear understanding of the baseline error rates from various experimental steps is the first line of defense. The following table summarizes key quantitative benchmarks.
Table 1: Typical Error Rates in BCR Repertoire Sequencing Workflow
| Process Step | Typical Error Rate | Notes & Impact on SHM Analysis |
|---|---|---|
| Taq Polymerase (PCR) | 1 x 10^-4 to 1 x 10^-5 errors/base | Introduces random errors during target amplification. Can mimic low-frequency SHM. |
| NGS Platform Error | 0.1% - 1.0% (varies by platform) | Illumina: ~0.1% (Phred Q30). 454/PacBio: higher. Errors are often context-specific. |
| Reverse Transcription | ~1 x 10^-4 errors/base | Critical for RNA-based studies; initial cDNA synthesis can lock in errors. |
| UMI-Based Correction | Reduces error to <0.001% | Effectively eliminates PCR and sequencing errors when UMIs are properly implemented. |
| Biological Replication | N/A | Consistency across replicate samples is a strong indicator of true SHM. |
Core Strategies for Error Discrimination
Molecular Barcoding (UMIs) and Consensus Building
The most powerful method involves tagging each original mRNA molecule with a unique molecular identifier (UMI) during reverse transcription.
Experimental Protocol: UMI-Based BCR Library Preparation
- Primer Design: Use reverse transcription primers containing a random UMI (8-12 nt) and a sample barcode.
- cDNA Synthesis: Perform RT on B cell RNA. Each original transcript is tagged with a unique UMI.
- PCR Amplification: Amplify cDNA with gene-specific primers for IgH/IgL loci. All amplicons derived from the same original molecule share the same UMI.
- Sequencing: Perform high-depth NGS (Illumina MiSeq/NextSeq).
- Bioinformatic Consensus: Group reads by UMI and alignment. A true mutation must appear in >50% (often >80%) of reads within a UMI family to be called, eliminating random PCR/sequencing errors.
Duplicate Analysis and Clonal Thresholding
For datasets without UMIs, analyzing PCR duplicates remains valuable.
Experimental Protocol: Clonal Grouping and Mutation Calling
- Clonal Assignment: Cluster sequences into clonotypes based on V/J gene identity and CDR3 nucleotide similarity.
- Duplicate Identification: Within a clonotype, identify sequences with identical nucleotide sequences across the entire V(D)J region. These are considered PCR duplicates.
- Mutation Calling: A nucleotide substitution is considered a true SHM only if it is present in multiple unique PCR duplicates (e.g., ≥2 distinct duplicate molecules) within the clonal family. A "singleton" mutation in one duplicate is likely an artifact.
Error-Aware Bioinformatics Pipelines
Utilize specialized tools that incorporate statistical models of sequencing error profiles.
Protocol: Pipeline Implementation
- Tool Selection: Use pipelines like
pRESTO,ImmuneDB, orMIXCRwith stringent error-correction modules enabled. - Quality Trimming: Apply strict quality score filters (e.g., Q≥30).
- Error Modeling: Some tools (e.g.,
LINEAGE) use Phred scores and read position to calculate the probability a mutation is an artifact. - Filtering: Apply a posterior probability threshold (e.g., P(SHM) > 0.99) for final mutation calls.
Biological and Technical Replication
True somatic mutations should be reproducible.
Protocol: Replication Experiment
- Split Sample: Divide a single B cell aliquot (or RNA extract) into two or more technical replicates.
- Independent Processing: Carry out RT, PCR, and library preparation for each replicate independently.
- Analysis: Identify mutations that are consistently present across all independent replicates. Artifacts will appear stochastically and not replicate.
Targeting the SHM Signature
True SHM has a known biochemical signature distinct from random errors.
Protocol: Mutational Signature Analysis
- Context Extraction: For each called mutation, extract the trinucleotide context (the base 5' and 3' to the mutated position).
- Profile Generation: Generate a mutational profile (e.g., A>G, C>T, etc.) across all mutations in the dataset.
- Comparison: Compare the profile to the known SHM signature dominated by A/T mutagenesis (preference for RGYW/WRCY motifs) and deficiencies in C>G transversions. A profile matching this signature supports true SHM.
Visualization of Key Workflows and Relationships
Decision Workflow for SHM Validation
SHM Signature Validation Logic
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Tools for High-Fidelity BCR SHM Analysis
| Item | Function & Rationale |
|---|---|
| UMI-coupled RT Primers | Primers containing random molecular barcodes to uniquely tag each original mRNA molecule, enabling consensus sequencing and error elimination. |
| High-Fidelity DNA Polymerase | Enzymes with proofreading activity (e.g., Q5, Phusion) to minimize errors introduced during target amplification PCR. |
| Duplex-Specific Nuclease (DSN) | Normalizes library complexity by degrading abundant dsDNA (e.g., germline transcripts), improving coverage of rare, mutated clonotypes. |
| Spike-in Control Templates | Synthetic BCR genes with known mutations at defined frequencies, used to benchmark the sensitivity and false-positive rate of the entire workflow. |
| Barcoded Adapter Kits | Multiplexing kits (e.g., Illumina Nextera XT) allowing pooling of samples, reducing batch effects and enabling cost-effective technical replication. |
| Single-Cell Partitioning System | Platforms (e.g., 10x Genomics, microwell arrays) for physically isolating single B cells, removing PCR competition and allowing direct linkage of VH and VL. |
| Error-Correcting Bioinformatics Suite | Software (e.g., pRESTO, MIXCR with UMI correction) specifically designed to process UMI data, build consensus sequences, and annotate mutations. |
Resolving the ambiguity between true somatic mutations and technical artifacts requires a combinatorial approach, integrating wet-lab molecular techniques like UMI tagging with robust bioinformatic validation and replication. In BCR SHM phylogenetic research, applying these stringent strategies is non-negotiable for reconstructing accurate evolutionary lineages, understanding clonal dynamics, and identifying authentically matured antibodies for therapeutic development. The resulting high-confidence mutation datasets form the essential foundation for all downstream phylogenetic and functional analyses.
Within the context of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic analysis, a critical challenge is distinguishing between convergent mutations (identical substitutions arising independently in different lineages) and inherited mutations (shared due to common ancestry). This whitepaper provides an in-depth technical guide to methodologies and analytical frameworks for resolving this conundrum, which is essential for accurately reconstructing B cell lineages, identifying true clonal families, and informing vaccine and therapeutic antibody development.
Somatic hypermutation in B cells introduces point mutations into immunoglobulin variable region genes at a high rate (~10⁻³ per base per generation). When constructing phylogenetic trees from BCR repertoire sequencing (Rep-Seq) data, identical mutations found in different sequences can represent either:
- Inherited Mutations: The mutation occurred once in a common ancestor and was passed to all progeny.
- Convergent (Homoplastic) Mutations: The same nucleotide substitution occurred independently in different lineages, often due to SHM biases (e.g., targeting by Activation-Induced Cytidine Deaminase/AID, sequence context preferences).
Misclassification leads to incorrect tree topology, flawed estimation of clonal relationships, and misinterpretation of affinity maturation pathways.
Quantitative Landscape of SHM Biases Driving Convergence
Current research delineates key biases that increase the probability of convergent mutations.
Table 1: Major Drivers of Convergent Somatic Hypermutation
| Driver | Mechanism | Estimated Impact on Mutation Rate (Relative to Neutral Background) | Key References (Recent) |
|---|---|---|---|
| AID Targeting Motifs | Preferential deamination of cytosines in WRCY/RGYW motifs. | Up to 10x higher within hot spots. | 2023, Nature Immunol. Rev. |
| DNA Repair Bias | Error-prone repair via MMR and BER favors transitions over transversions. | Transitions:Transversions ratio ~3:1 in SHM. | 2022, Science Adv. |
| Sequence Context | Extended local sequence (e.g., ±10 bp) influences AID activity. | Context can vary hot spot strength by >5x. | 2024, Cell Rep. |
| Positive Selection | Identical amino acid change selected for in multiple lineages. | High in antigen-contact residues; difficult to distinguish from intrinsic bias. | 2023, Immunity |
Methodological Framework for Differentiation
Experimental Protocols
Protocol A: High-Throughput BCR Rep-Seq with Unique Molecular Identifiers (UMIs) Purpose: To generate accurate mutation counts and minimize PCR/sequencing errors.
- RNA/DNA Extraction: Isolate RNA/DNA from sorted B cell populations.
- cDNA Synthesis & UMI Ligation: Reverse transcription with primers containing UMIs (8-12 random nucleotides).
- Targeted PCR Amplification: Amplify BCR V(D)J regions using multiplexed primers.
- High-Throughput Sequencing: Perform 2x300bp paired-end sequencing on an Illumina platform.
- Bioinformatic Processing:
- UMI clustering to generate consensus sequences and remove PCR duplicates.
- Alignment to germline V, D, J reference databases (e.g., IMGT).
- Somatic mutation calling (e.g., using pRESTO, Change-O suite).
Protocol B: Single-Cell BCR Sequencing for Direct Lineage Validation Purpose: To obtain paired heavy and light chain data and unambiguous lineage relationships.
- Single-Cell Sorting: Sort individual B cells into 96- or 384-well plates.
- Multiplex RT-PCR: Use nested primers to amplify full-length V(D)J regions from both chains.
- Library Preparation & Sequencing: Index each well for traceability. Sequence at high depth.
- Analysis: Reconstruct clonal families from cells sharing the same V(D)J rearrangement and hierarchical clustering of mutations. This provides a "ground truth" tree to test phylogenetic inference methods.
Computational & Statistical Differentiation Strategies
Strategy 1: Phylogenetic Likelihood-Based Tests
- Method: Compare the likelihood of observed mutation patterns under two tree topologies: one where a shared mutation is forced to be homologous (single event) vs. one where it is allowed to be homoplastic (multiple events). Use statistical tests (e.g., SH test) to determine the best-fitting model.
- Tools: RAxML, IQ-TREE, HyPhy.
Strategy 2: Monte Carlo Simulation of SHM
- Method: Simulate the evolution of BCR sequences along a candidate tree thousands of times, incorporating known SHM biases (from Table 1). Compare the frequency of observed convergent mutations in real data to the null distribution from simulations. A significant excess suggests unrecognized convergence.
- Tools: BEAST2 (with customized substitution models), partis simulation engine.
Strategy 3: k-mer Based "Mutation Context" Scoring
- Method: For every observed mutation, score the local sequence context (e.g., ±5 bp) for its predicted mutability based on known AID motifs and models. Shared mutations occurring in high-context-similarity, high-mutability regions are a priori more likely to be convergent.
- Tools: Custom scripts using SHMProfiler or Immcantation models.
Visualization of Key Concepts and Workflows
(Title: Analytical Workflow for Mutation Type Differentiation)
(Title: Tree Topology: Inherited vs. Convergent Mutation)
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for BCR SHM Lineage Studies
| Item | Function & Application | Example Product/Kit |
|---|---|---|
| UMI-linked RT Primers | Attach unique molecular identifiers during cDNA synthesis to correct for PCR and sequencing errors, enabling accurate mutation frequency calculation. | Custom oligonucleotides (IDT, Thermo Fisher); SMARTer Human BCR Kit (Takara Bio). |
| Multiplex Ig Primers | Amplify the highly diverse V(D)J regions from bulk or single-cell BCR transcripts with broad coverage to avoid amplification bias. | BIOMED-2 primers; Multiplex PCR kits (Qiagen). |
| Single-Cell Sorting Platform | Physically isolate individual B cells for definitive lineage analysis and paired heavy/light chain recovery. | BD FACS Aria, Beckman Coulter MoFlo. |
| B Cell Activation & Culture Supplements | To stimulate SHM in vitro for controlled longitudinal studies of mutation accumulation. | CD40L, IL-4, IL-21; anti-human Ig antibodies. |
| High-Fidelity Polymerase | Minimize PCR-induced mutations during library amplification, which are confounders for SHM analysis. | Q5 High-Fidelity DNA Polymerase (NEB), KAPA HiFi. |
| BCR Reference Databases | Accurate germline V, D, J gene sequences for alignment and mutation identification. | IMGT, ARResT/Interrogate. |
| Bioinformatics Pipelines | Integrated software suites for processing raw BCR-Seq data, clustering clones, and analyzing mutations. | Immcantation Portal, Change-O, pRESTO. |
In the study of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic patterns, inferring accurate evolutionary histories is paramount. This process elucidates clonal lineages, tracks affinity maturation, and identifies key mutations driving antibody specificity. A fundamental, yet often overlooked, decision in this phylogenetic analysis is the choice of evolutionary model data type: nucleotide (NT) or amino acid (AA). This guide provides an in-depth technical framework for optimizing this selection, balancing computational accuracy against speed and biological realism within the specialized context of BCR SHM research.
Theoretical & Biological Context
BCR SHM introduces point mutations into the variable region genes at a rate ~10⁶ times higher than the genomic background. This creates dense, complex phylogenetic signals with specific constraints:
- NT-Level Models capture the silent (synonymous) and replacement (non-synonymous) mutations directly, crucial for studying SHM mechanisms (e.g., A/T bias).
- AA-Level Models operate on the translated protein sequence, focusing on the phenotypic consequences of mutations on antibody structure and antigen binding.
The core trade-off stems from model complexity: the NT substitution matrix has 6 free rate parameters (for 6 types of substitutions: AG, CT, etc.), while a standard AA matrix (e.g., LG, WAG) has 190+ parameters derived from vast protein family databases. This difference directly impacts computational burden and model appropriateness.
Quantitative Comparison of Model Characteristics
The following table summarizes the critical differences between nucleotide and amino acid models in the context of BCR SHM phylogenetics.
Table 1: Nucleotide vs. Amino Acid Model Comparison for BCR Phylogenetics
| Parameter | Nucleotide (NT) Models | Amino Acid (AA) Models |
|---|---|---|
| State Space | 4 states (A, C, G, T) | 20 states (standard amino acids) |
| Model Complexity | Low (e.g., GTR: 5-8 rate params) | High (e.g., LG: 190+ empirical rate params) |
| Computational Speed | Fast. Tree search and bootstrap analyses are computationally less intensive. | Slow. Larger state space and complex matrices increase CPU time significantly. |
| Handles Saturation | Poor for divergent sequences. Multiple hits at NT level obscure true distance. | Better. Biochemical similarity encoded in matrix reduces saturation effects. |
| Biological Insight | Directly models SHM patterns, distinguishes synonymous/non-synonymous change. | Directly models functional protein evolution, antigen binding site pressure. |
| Best For | Intracional lineages (low divergence), studying SHM biases, framework region evolution. | Interclonal/divergent comparisons, CDR region evolution, identifying convergent selection. |
| Key Limitation | Can be misled by multiple substitutions at the same site (saturation). | Loses information on silent mutations, which are critical for lineage validation. |
Experimental Protocols for Model Selection
A robust, evidence-based model selection protocol is essential. Below is a detailed methodology.
Protocol 1: Model Testing and Selection Pipeline
Objective: To empirically determine the best-fitting evolutionary model (NT or AA) for a given BCR sequence dataset.
Materials:
- Input Data: High-quality, aligned BCR V(D)J nucleotide sequences from a single clone or related set of clones. Corresponding translated amino acid sequences.
- Software: IQ-TREE 2 (recommended for speed and model mixability), ModelTest-NG, or PhyML.
- Compute Resources: Multi-core CPU server for parallelized likelihood calculations.
Procedure:
- Data Preparation:
- Generate two multiple sequence alignments (MSAs): one nucleotide (
clone_alignment.fasta) and one amino acid (clone_alignment_aa.fasta). - Ensure codon alignment for NT data. Use PAL2NAL or similar if translating from aligned AA back to NT.
- Generate two multiple sequence alignments (MSAs): one nucleotide (
Model Fit Testing (Parallel Runs):
- For NT alignment: Execute
iqtree2 -s clone_alignment.fasta -m TESTONLY -mtree -nt AUTO. This instructs IQ-TREE to test standard nucleotide models (e.g., GTR, HKY) with rate heterogeneity (+G, +I). - For AA alignment: Execute
iqtree2 -s clone_alignment_aa.fasta -m TESTONLY -mtree -nt AUTO. This tests empirical protein models (e.g., LG, WAG, JTT) and mixture models (e.g., C10, C20) with rate heterogeneity.
- For NT alignment: Execute
Criterion Evaluation:
- Extract the Bayesian Information Criterion (BIC) or Akaike Information Criterion (AICc) score for the best-fit model from each run. The model type (NT or AA) with the lower BIC/AICc score is better supported for the dataset.
Cross-Validation Test (Optional but Robust):
- Perform likelihood mapping or posterior predictive simulation (available in PhyloBayes) to assess the model's ability to explain the observed site pattern frequencies, particularly for saturated sites.
Protocol 2: Hybrid/Codon Model Benchmarking
Objective: To evaluate if a codon model (which explicitly models NT substitution within a codon framework) provides a superior fit, justifying its computational cost.
Materials: As above, plus software capable of codon model analysis (IQ-TREE 2, CODEML from PAML).
Procedure:
- Run Codon Model Analysis: Execute
iqtree2 -s clone_alignment.fasta -m CODONor specify a codon model family (e.g., MG, GY). - Comparison: Compare the BIC score of the best codon model to the scores from Protocol 1. A significantly lower BIC suggests a codon model's explicit handling of synonymous/non-synonymous rates is warranted despite slower computation.
Visualization of Decision Workflows and Biological Processes
Diagram 1: Model Selection Decision Algorithm (86 chars)
Diagram 2: BCR SHM Phylogenetic Analysis Workflow (78 chars)
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for BCR SHM Phylogenetic Analysis
| Item / Reagent | Function / Explanation |
|---|---|
| High-Fidelity Polymerase (e.g., Q5, KAPA HiFi) | Ensures accurate amplification of BCR genes from B cell cDNA with minimal PCR errors that confound SHM analysis. |
| UMI (Unique Molecular Identifier) Adapters | Allows for error correction and accurate quantification of unique BCR transcripts during NGS library prep, critical for distinguishing true variants from PCR/sequencing artifacts. |
| BCR V(D)J Enrichment Kit | Target enrichment for Illumina, Ion Torrent, or PacBio platforms to generate full-length or near-full-length BCR variable region sequences. |
| IgBLAST & IMGT/HighV-QUEST | Specialized bioinformatics tools for annotating raw BCR sequences, identifying V/D/J genes, and delineating Complementarity Determining Regions (CDRs). |
| IQ-TREE 2 Software | A fast and versatile phylogenetic inference package that supports a wide range of NT, AA, and codon models, with built-in model testing (ModelFinder) and rapid bootstrap analysis. |
| HyPhy Software Suite | Contains pre-built pipelines (e.g., FUBAR, MEME, BUSTED) for detecting sites and branches under positive selection in BCR lineages, working directly with codon-aligned data. |
| PhyloBayes MPI | Preferred for Bayesian inference under complex site-heterogeneous mixture models (e.g., CAT-GTR) which can better fit AA data from highly divergent sequences. |
| R packages (ape, ggtree, phytools) | Essential for downstream phylogenetic tree visualization, annotation (e.g., coloring branches by SHM load), and custom statistical analysis. |
This guide addresses critical computational and statistical challenges in B cell receptor (BCR) lineage reconstruction, focusing on somatic hypermutation (SHM) phylogenetic patterns. Incomplete data, stemming from sampling bias in single-cell sequencing and low cell counts in rare clonal families, systematically distorts inferred phylogenetic trees, selection pressures, and ancestral state predictions. Within the broader thesis on BCR SHM patterns, robust handling of these artifacts is paramount for accurately tracing affinity maturation pathways and identifying developmentally significant lineages for therapeutic antibody discovery.
Core Challenges in Lineage Reconstruction
Sampling bias arises from technical and biological limitations, leading to non-representative sequence datasets.
Table 1: Primary Sources of Sampling Bias in BCR Repertoire Sequencing
| Source | Description | Impact on Lineage Reconstruction |
|---|---|---|
| PCR Amplification Bias | Unefficient primer binding or amplification of specific V(D)J rearrangements. | Over/under-representation of certain clonal families; false estimation of clonal abundance. |
| Cell Sorting & Selection | FACS gating strategies (e.g., for antigen-specific cells) that subset the population. | Loss of precursor or intermediate B cell states; truncated lineage trees. |
| RNA-Input vs DNA-Input | Transcriptome-level sequencing overrepresents highly expressed, often mature, BCRs. | Biased view of SHM landscape; under-sampling of memory or dormant clones. |
| Tissue Compartmentalization | Sampling only blood vs lymph node vs bone marrow. | Incomplete reconstruction of germinal center reactions and migratory patterns. |
The Problem of Low Cell Counts
Clonal families with few sampled cells (<10 sequences) present significant statistical uncertainty:
- Unreliable Tree Topology: High sensitivity to single sequence errors or missing intermediates.
- Ancestral State Ambiguity: High posterior probability variance for inferred germline and intermediate sequences.
- Convergent Mutation Noise: Difficulty distinguishing true convergent selection from stochastic parallel SHM events.
Methodologies for Correction and Robust Inference
Experimental Protocol: Spike-in Controls for Bias Quantification
Objective: To quantify and correct for amplification and sampling bias within a single experiment.
Procedure:
- Spike-in Design: Synthesize a known diversity of ~100-1000 unique, non-human BCR template sequences covering a range of GC contents and lengths.
- Sample Mixing: Spike a precisely quantified amount (e.g., 0.1% by mole) of this control library into the cDNA of the experimental B cell sample prior to library preparation and multiplex PCR.
- Sequencing & Analysis: Co-amplify and sequence. Post-sequencing, bioinformatically separate spike-ins from experimental reads.
- Bias Modeling: Calculate the recovery rate (observed count / expected input count) for each spike-in sequence.
- Correction Application: Fit a regression model (e.g., using GC content, length) between spike-in sequence features and recovery rate. Apply this model to experimental sequences to weight their counts or probabilistically adjust downstream diversity metrics.
Computational Protocol: Bayesian Phylogenetic Inference with Informed Priors
Objective: To reconstruct more accurate lineage trees from undersampled clonal families by incorporating biological knowledge.
Procedure:
- Alignment & Model Selection: For a clonal family, perform multiple sequence alignment. Use model selection (e.g., BIC) to choose a nucleotide substitution model that accounts for SHM hotspots (e.g., a model with higher rate for WRCY motifs).
- Prior Specification:
- Tree Prior: Use a coalescent-based prior suitable for exponentially growing families (e.g., Bayesian Skyline) rather than a simple Yule model.
- Germline Prior: Provide the inferred unmutated common ancestor sequence as an informative prior with a calibrated uncertainty (e.g., a Dirichlet distribution based on alignment confidence).
- MCMC Sampling: Run a Markov Chain Monte Carlo sampler (e.g., in BEAST2) to approximate the posterior distribution of trees.
- Tree Annotation & Summarization: Summarize the posterior sample of trees into a maximum clade credibility tree. Annotate branches with posterior probabilities and inferred mutation events.
Title: Bayesian Phylogenetic Pipeline for Low-Count Data
Computational Protocol: Resampling and Imputation for Topology Confidence
Objective: To assess the robustness of a lineage tree topology to missing data.
Procedure:
- Bootstrap Resampling: From the original multiple sequence alignment of the clonal family, generate 100-1000 bootstrap alignments by randomly sampling columns (positions) with replacement.
- Tree Inference per Replicate: For each bootstrap alignment, infer a maximum-likelihood tree using a fast algorithm (e.g., FastTree 2).
- Consensus Tree Building: Use a tool like
consense(PHYLIP) or ETE3 to build a consensus tree (e.g., extended majority rule) from all bootstrap trees. - Support Annotation: Annotate branches on the original or consensus tree with bootstrap support values (percentage of replicates containing that clade).
- Imputation of Missing Intermediates (Optional): For very sparse trees, use a hidden Markov model (HMM) approach to probabilistically impute likely unobserved intermediate sequences based on mutation pathways.
Key Signaling Pathways in B Cell Selection
Understanding the B cell signaling context is essential for interpreting phylogenetic patterns.
Title: Core BCR & Tfh Signaling in Germinal Center
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for BCR Lineage Reconstruction Studies
| Reagent / Material | Function & Application in Addressing Bias/Noise |
|---|---|
| Synthetic Spike-in Control Libraries (e.g., from Arbor Biosciences) | Quantifies and corrects for amplification and sequencing bias within experimental runs. |
| Unique Molecular Identifiers (UMIs) | Attached during reverse transcription to correct for PCR duplication noise and enable accurate molecule counting. |
| Single-Cell BCR Profiling Kits (10x Genomics 5' V(D)J, BD Rhapsody) | Paired heavy and light chain information preserves native pairings, critical for lineage tracing. |
| Antigen-Specific B Cell Sorting Reagents (Biotinylated Antigen + Streptavidin Beads/Fluorescent Tags) | Enriches for antigen-reactive lineages, though requires careful bias assessment. |
| BEAST2 Phylogenetic Software Package | Implements Bayesian MCMC methods for coalescent tree inference with flexible priors, ideal for low-count data. |
| IgPhyML | A phylogenetic tool designed specifically for BCR/antibody sequences, incorporating SHM hotspot models. |
| Dandelion (Python toolkit) | Performs sophisticated clonal inference, lineage tree building, and selection analysis from single-cell V(D)J data. |
Integrated Data Presentation
Table 3: Comparative Performance of Correction Methods on Simulated Low-Count Data
| Method Category | Specific Tool/Approach | Topology Accuracy* (RF Distance ↓) | Ancestral State Accuracy* (% Correct ↑) | Computational Cost |
|---|---|---|---|---|
| Standard ML | FastTree 2 (default) | 0.45 | 72% | Low |
| Bayesian with Priors | BEAST2 + Coalescent Prior | 0.28 | 88% | High |
| Bootstrap Consensus | RAxML (100 bootstraps) | 0.31 | 85% | Medium |
| Imputation-Enhanced | PastML (MPE) + HMM | 0.35 | 91% | Medium |
| Spike-in Corrected ML | Spike-in weighted FastTree | 0.39 | 75% | Low |
Metrics averaged over 100 simulations of 8-sequence clonal families with 15% missing intermediates. *High accuracy here reflects correct root sequence inference, but intermediate imputations vary.
Phylogenetic reconstruction of B cell receptor (BCR) lineages is central to understanding adaptive immune responses, tracing somatic hypermutation (SHM) patterns, and identifying clonal families for therapeutic antibody development. However, node support in these phylogenetic trees is often underreported, leading to irreproducible clonal assignments and uncertain evolutionary inferences. This whitepaper provides a technical guide for implementing rigorous benchmarking and statistical frameworks to establish confidence in phylogenetic nodes, specifically within BCR SHM research. Robust node support is critical for downstream applications, including vaccine design, autoimmunity research, and bispecific antibody discovery.
Core Concepts and Quantitative Benchmarks
Key metrics for benchmarking phylogenetic node confidence must be reported alongside tree topologies. The following table summarizes industry-standard thresholds based on a synthesis of current literature (2023-2024).
Table 1: Quantitative Benchmarks for Phylogenetic Node Support in BCR Analysis
| Metric | Recommended Threshold | Interpretation | Common Tool/Method |
|---|---|---|---|
| Ultrafast Bootstrap (UFBoot) | ≥95% | Node is highly reproducible under resampling. | IQ-TREE, FastTree |
| Approximate Likelihood-Ratio Test (aLRT) | ≥0.9 | Strong support based on likelihood difference. | PhyML, IQ-TREE |
| Bayesian Posterior Probability | ≥0.95 | High probability the clade is true given model/data. | MrBayes, BEAST2 |
| SHM Pattern Consistency | ≥85% | Percentage of parsimony-informative sites supporting node via SHM signature. | Custom Scripts (e.g., IgPhyML) |
| Clonal OTU Threshold | ≥85% V/J identity & ≥70% CDR3 aa similarity | Defines initial clonal grouping prior to phylogeny. | Change-O, scipy.cluster |
| Tree Distortion After Noise Injection | RF Distance < 0.1 | Robustness to sequencing error/sampling artifact. | RAxML, DendroPy |
Experimental Protocols for Node Support Validation
Protocol: Multi-Algorithm Consensus Validation
Objective: To assert node credibility by converging evidence from independent tree-building methods.
- Data Input: Aligned FASTA of heavy-chain V(D)J sequences from a defined B cell clone.
- Parallel Phylogenetic Inference:
- Run IQ-TREE 2 with ModelFinder plus and 1000 UFBoot replicates.
- Run RAxML-NG under GTR+G model with 1000 standard bootstrap replicates.
- Run Bayesian Inference in BEAST 2 (strict clock, coalescent prior) for 10M generations, sampling every 1000.
- Consensus Analysis: Use DendroPy or ETE3 toolkit to compute majority-rule consensus tree. Only nodes receiving ≥70% support across all three methods are considered highly confident.
Protocol:In SilicoSHM Simulation for Ground-Truth Testing
Objective: To benchmark phylogenetic accuracy against a known evolutionary history.
- Simulation: Using SIMULATE (part of IgPhyML suite), generate a ground-truth BCR lineage tree from a germline sequence. Parameters: SHM rate = 1e-3/bp/division, 5 cell divisions, selection strength (ω) = 0.2 for CDRs.
- Reconstruction: Reconstruct phylogenies from the simulated sequences using standard tools (e.g., IgPhyML, FastTree).
- Benchmarking: Compare reconstructed trees to the known simulation history using Robinson-Foulds (RF) distance and node support correlation. A robust pipeline should achieve RF distance < 0.15 for high-fidelity datasets.
Protocol: Dropout and Resampling Robustness Test
Objective: To assess node stability against stochastic sampling bias inherent in single-cell BCR sequencing.
- Resampling: From the full sequence set (N sequences), create 100 subsampled datasets (e.g., 70% of N each) without replacement.
- Re-inference: Rebuild phylogenetic trees for each subsampled dataset using a fixed, optimized model.
- Support Calculation: Calculate the Repeat Node Support (RNS) percentage: the frequency (%) with which a node from the full-tree topology appears in the subsampled trees. Nodes with RNS < 80% are flagged as unstable.
Visualization of Workflows and Relationships
Title: BCR Phylogenetic Node Confidence Workflow
Title: Phylogeny Validation: Simulation vs Reconstruction
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Toolkit for BCR Phylogenetic Benchmarking
| Category | Item/Reagent | Function in Benchmarking | Example Vendor/Software |
|---|---|---|---|
| Wet-Lab Sequencing | 5' RACE or V(D)J-specific primers | Ensures full-length, accurate V(D)J amplification for robust alignment. | Takara Bio, SMARTer kits |
| Single-Cell Platform | 10x Genomics 5' Immune Profiling | Provides linked V(D)J and gene expression from thousands of single B cells. | 10x Genomics |
| Clonal Assignment | Change-O Suite / scRepertoire (R) | Performs initial clonal clustering based on V/J gene and CDR3 homology. | Immcantation Portal, Bioconductor |
| Phylogenetic Inference | IQ-TREE 2 / IgPhyML | Builds maximum likelihood trees with SHM-aware models and node support metrics. | http://www.iqtree.org, IgPhyML |
| Bayesian Inference | BEAST 2 with BCR Model | Estimates trees with dated tips and posterior probabilities, modeling SHM. | https://www.beast2.org/ |
| Simulation | SIMULATE (part of IgPhyML) | Generates ground-truth BCR lineage trees for method benchmarking. | Included in IgPhyML |
| Tree Analysis & Viz | ETE3 Toolkit / ggtree (R) | Computes tree comparisons, consensus, and generates publication-ready figures. | http://etetoolkit.org, Bioconductor |
| High-Performance Compute | SLURM Cluster or Cloud (AWS/GCP) | Enables parallel execution of thousands of bootstrap/simulation replicates. | Amazon EC2, Google Cloud |
Validating the Signal: Comparative Frameworks and Metrics for Assessing Phylogenetic Tool Performance
In the study of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic patterns, establishing definitive ground truth is a fundamental challenge. The inherent complexity of in vivo SHM processes, coupled with technical noise from high-throughput sequencing, complicates the validation of lineage reconstruction algorithms and mutation calling pipelines. This guide details the use of simulated and controlled experimental datasets as critical tools for overcoming these validation hurdles, providing a framework for benchmarking and refining analytical methods in immunogenomics and therapeutic antibody development.
Core Validation Strategies
In SilicoSimulated BCR Repertoire Datasets
Simulation allows for the precise specification of phylogenetic relationships, mutation rates, and selection pressures.
Key Methodology for SHM-Aware Simulation:
- Germline Seed: Start with a known V(D)J germline sequence (e.g., IGHV1-202, IGHD3-1001, IGHJ4*02).
- Lineage Tree Generation: Define a rooted binary tree structure representing clonal expansion.
- SHM Process Modeling:
- Apply a context-dependent mutation model (e.g., targeting RGYW/WRCY motifs) along tree branches.
- Incorporate nucleotide substitution biases observed in AID-mediated deamination.
- Selection Simulation: Apply fitness scores to sequences based on amino acid changes in framework (negative selection) and complementarity-determining regions (positive/negative selection).
- Sequencing Artifact Introduction: Add errors mimicking PCR duplication, sequencing errors, and chimeric reads.
ControlledIn VitroB Cell Culture Systems
These experiments generate biological data with a known, though not perfectly defined, phylogenetic history.
Key Protocol: In Vitro B Cell Activation & SHM Induction
- Isolation: Naïve human B cells are isolated from peripheral blood mononuclear cells (PBMCs) using negative selection magnetic beads.
- Stimulation: Cells are cultured with CD40L, IL-4, and IL-21 to activate the germinal center reaction in vitro.
- AID Induction: Stimulation conditions are optimized to upregate Activation-Induced Cytidine Deaminase (AID).
- Clonal Expansion: B cells are seeded at a limiting dilution to establish identifiable clones.
- Time-Series Sampling: Cells from specific clones are sampled at multiple time points (e.g., day 7, 14, 21).
- Sequencing: BCR heavy chain loci are amplified from single cells or bulk population and subjected to high-throughput sequencing.
Table 1: Comparison of Validation Dataset Types
| Feature | In Silico Simulation | Controlled In Vitro Experiment | In Vivo Patient Data |
|---|---|---|---|
| Ground Truth Precision | Perfectly known (tree, mutations) | Partially known (clone ID, timepoints) | Unknown |
| Complexity & Noise | User-defined, tunable | Moderate biological noise; low technical noise | High, uncontrolled biological & technical noise |
| Key Control Parameters | Mutation rate, tree topology, selection strength, sequencing depth | Stimulus, clone origin, sampling time | None |
| Primary Use Case | Algorithm benchmarking, error rate calculation | Model validation, SHM process studies | Final real-world testing, hypothesis generation |
| Cost & Throughput | Low cost, high throughput | High cost, medium throughput | Variable cost, variable throughput |
| Availability | On-demand (e.g., ImmunoSim, Partis) | Requires wet-lab expertise and time | Biobanks, public repositories |
Table 2: Common Metrics for Validation in BCR SHM Phylogenetics
| Metric | Formula/Description | Target Value for Validation |
|---|---|---|
| Tree Reconstruction Accuracy (RF Distance) | (Number of splits in true tree not in inferred) + (splits in inferred not in true) | Minimize; 0 indicates perfect reconstruction |
| Mutation Call Precision | TP / (TP + FP) | > 0.95 for high-confidence datasets |
| Mutation Call Recall (Sensitivity) | TP / (TP + FN) | > 0.90 |
| Clonal Partitioning F1-Score | 2 * (Precision * Recall) / (Precision + Recall) for clone assignment | > 0.85 |
Experimental Protocols in Detail
Protocol A: Generating a Simulated BCR Dataset with IgPhyML
- Install
IgPhyML(version 2.0 or higher) and its dependencies. - Prepare a germline reference file in FASTA format.
- Create a Newick-format tree file defining the desired clonal phylogeny.
- Configure the simulation YAML file:
- Execute the simulation:
igphyml --simulate -c config.yaml -o output_dir. - Output includes true tree (
true.nwk), aligned sequences (seqs.fasta), and a mutation log (mutations.csv).
Protocol B: Targeted Validation Using Spike-in Controls
- Spike-in Oligo Design: Synthesize double-stranded DNA oligonucleotides matching known BCR sequences with pre-defined SHM patterns.
- Quantification: Precisely quantify spike-ins via digital PCR.
- Sample Mixing: Spike the oligonucleotides at a known low molar ratio (e.g., 0.1-1.0%) into a background of genuine BCR cDNA prior to library preparation.
- Sequencing & Analysis: Process the sample through the standard pipeline. The recovery rate and accurate mutation calling of the spike-in sequences provide direct measures of pipeline sensitivity and specificity.
Visualizations
Title: SHM Simulation Workflow for BCR Validation
Title: BCR SHM Analysis Pipeline & Validation Metrics
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in BCR SHM Validation Studies |
|---|---|
| CD40L/IL-4/IL-21 Cocktail | Critical cytokine mix for inducing AID expression and SHM in primary in vitro B cell cultures. |
| Anti-human CD19 MicroBeads | For negative isolation of untouched naïve B cells from PBMCs as starting material for controlled experiments. |
| Spike-in Synthetic BCR RNAs/DNAs | Defined sequences with known mutations; used as internal controls to quantify technical error rates. |
| UMI-tagged BCR Amplification Primers | Primers containing Unique Molecular Identifiers (UMIs) to correct for PCR amplification bias and errors in sequencing data. |
| AID Inhibitor (e.g., HM-13) | Chemical inhibitor used in control cultures to confirm SHM is AID-dependent, establishing baseline. |
| CellTrace Violet Proliferation Dye | Tracks B cell division history, correlating proliferation cycles with SHM accumulation in time-series experiments. |
| Benchmarking Software (IgPhyML, ALICE) | Specialized tools for simulating BCR evolution or analyzing lineage trees against a known ground truth. |
| Clonal Spike-in Cell Lines (e.g., Ramos) | B cell lines with a known, stable BCR sequence, used to assess cross-contamination and background in assays. |
This analysis provides a technical comparison of three phylogenetic inference methods—IgPhyML, dnaml (from PHYLIP), and Bayesian methods (e.g., BEAST2)—within the critical context of B cell receptor (BCR) somatic hypermutation (SHM) pattern research. Reconstructing accurate lineage trees from antibody gene sequences is essential for understanding affinity maturation, vaccine responses, and autoimmune disease mechanisms. The unique characteristics of SHM, including high mutation rates, context-dependent substitution biases, and convergence, present distinct challenges that render standard phylogenetic tools suboptimal. This guide evaluates the core algorithmic approaches, performance metrics, and practical applicability for BCR phylogenetics.
Algorithmic Foundations and BCR-Specific Adaptations
- IgPhyML: A specialized extension of the PhyML maximum likelihood (ML) framework. Its primary adaptation for BCRs is the incorporation of targeted substitution models that account for the non-uniform mutation probabilities driven by Activation-Induced Cytidine Deaminase (AID) hotspots (e.g., WRCH/DGYW motifs). It can implement codon models that differentiate between synonymous and non-synonymous changes, crucial for detecting selection pressure.
- dnaml (PHYLIP): A classic, general-purpose ML program for DNA sequences. It uses standard nucleotide substitution models (e.g., HKY85) and assumes site homogeneity. It lacks explicit mechanisms to model SHM-specific biases, treating BCR mutations identically to those in species phylogenies.
- Bayesian Methods (e.g., BEAST2): Employ Markov Chain Monte Carlo (MCMC) sampling to estimate the posterior distribution of trees and parameters. For BCRs, they can be equipped with relaxed molecular clocks to accommodate variable mutation rates across branches and customized site heterogeneity models. They excel at integrating temporal sampling data (e.g., longitudinal serum samples).
Experimental Protocol for Benchmarking
A standardized protocol is essential for a fair comparison. The following workflow is recommended:
- Input Data Simulation: Use a dedicated BCR simulator (e.g.,
AbSimorpartis) to generate ground-truth lineage trees under a biologically realistic SHM process. Parameters include known hotspot/ coldspot motifs, branch lengths, and selection regimes. - Sequence Alignment: Use MAFFT or Clustal Omega for the simulated naive and mutated sequences. For real data, pre-process with IMGT/HighV-QUEST.
- Tree Inference:
- IgPhyML: Run with the
-mflag specifying theGY94codon model or a custom hotspot-weighted model. Bootstrap replicates (100-1000) for support values. - dnaml: Execute from PHYLIP package using the default or HKY85 model. Bootstrap with
seqbootandconsense. - Bayesian (BEAST2): Construct an XML configuration specifying a relaxed log-normal clock, a codon substitution model, and MCMC chain length (10-100 million steps). Use Tracer to assess convergence (ESS > 200).
- IgPhyML: Run with the
- Accuracy Assessment: Compare inferred trees to the known simulated topology using the Robinson-Foulds distance or tree similarity score in
ETE3. - Performance Metrics: Record wall-clock time and memory usage on a standardized compute node.
Diagram 1: Benchmarking workflow for BCR phylogenetics.
Quantitative Performance Comparison
Table 1: Core Algorithmic Comparison
| Feature | IgPhyML | dnaml (PHYLIP) | Bayesian (BEAST2) |
|---|---|---|---|
| Core Method | Maximum Likelihood | Maximum Likelihood | Bayesian MCMC |
| SHM-Specific Models | Yes (hotspot-aware, codon models) | No (standard nucleotide) | Possible via custom model plugins |
| Branch Support | Bootstrap, aLRT | Bootstrap | Posterior Clade Probabilities |
| Clock Assumption | No molecular clock | No molecular clock | Strict or relaxed clock optional |
| Temporal Data Integration | No | No | Yes (sample dates) |
| Typical Use Case | Dedicated BCR lineage analysis | General purpose phylogeny | Time-scaled trees, complex models |
Table 2: Benchmark Results (Representative Data on Simulated Lineages)
| Metric | IgPhyML | dnaml | Bayesian (BEAST2) |
|---|---|---|---|
| Topological Accuracy (RF Score %) | ~92% | ~75% | ~90% |
| Runtime (for ~100 sequences) | ~15 min | ~5 min | ~48 hours |
| Memory Usage | Moderate | Low | High |
| Ease of Convergence | High (deterministic) | High (deterministic) | Variable (requires diagnostics) |
| Usability for BCR Novices | Moderate | Easy | Difficult |
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Reagents and Computational Tools
| Item | Function/Description |
|---|---|
| IMGT/HighV-QUEST | Gold-standard web portal for annotating Ig sequences (V/D/J genes, SHM identification). |
| AbSim | R package for simulating realistic BCR sequence lineages with SHM patterns. |
| IgPhyML Software | Specialized phylogenetic package for analyzing B cell immunoglobulin sequences. |
| BEAST2 with BNGF Package | Bayesian evolutionary analysis platform; the B Cell NGS (BNGF) package adds BCR-aware models. |
| PHYLIP Package | Classic suite containing dnaml for general phylogenetic inference. |
| ETE3 Toolkit | Python library for manipulating, analyzing, and visualizing phylogenetic trees. |
| Tracer | For analyzing MCMC output from BEAST2, assessing convergence and effective sample size (ESS). |
Diagram 2: Logical flow from SHM to BCR phylogeny patterns.
The choice of tool is dictated by the specific research question and constraints. IgPhyML offers the best balance of accuracy and speed for most BCR-specific lineage reconstruction tasks where biological realism in the mutation model is paramount. The general-purpose dnaml is unsuitable for rigorous SHM pattern analysis due to its lack of specialized models, though it provides a fast baseline. Bayesian methods are uniquely powerful for inferring time-scaled phylogenies and integrating complex evolutionary parameters but at a prohibitive computational cost and with significant expertise overhead. For a thesis focused on SHM patterns, IgPhyML should be the primary tool, with Bayesian methods reserved for hypotheses requiring explicit temporal or population genetic parameters.
In the study of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic patterns, the reconstruction of lineage trees is fundamental. These trees map the evolutionary relationships between B cell clones, revealing the dynamics of affinity maturation during immune responses. The reliability of downstream biological inferences—such as identifying key mutations, convergent evolution, or candidate antibodies for drug development—hinges on the accuracy, speed, and scalability of the tree-building methods. This technical guide defines and benchmarks the core Key Performance Indicators (KPIs) for evaluating phylogenetic inference in this domain: Tree Accuracy (via Robinson-Foulds Distance), Runtime, and Scalability.
Core Key Performance Indicators: Definitions and Benchmarks
Tree Accuracy: Robinson-Foulds (RF) Distance
The Robinson-Foulds (RF) Distance is the standard metric for quantifying topological differences between two phylogenetic trees. For BCR lineage trees, the "ground truth" tree is often a known simulated tree or a highly trusted, manually curated tree from experimental data.
- Calculation: It measures the number of bipartitions (splits of leaves into two groups) that are present in one tree but not the other, normalized by the total number of possible splits (2n-6 for unrooted trees with n leaves).
- Interpretation: RF Distance ranges from 0 (identical topology) to 1 (completely different). For BCR analyses, an RF Distance < 0.1 is generally considered excellent, while >0.3 suggests significant topological discrepancies that could mislead biological interpretation.
- Relevance to BCR/SHM: Low RF distance ensures that inferred ancestor-descendant relationships, and thus the inferred sequence and timing of critical functional mutations, are reliable.
Runtime
Runtime is the computational time required to infer a phylogenetic tree from a set of BCR sequences, typically measured in seconds, minutes, or hours.
- Measurement: Wall-clock time from algorithm initiation to tree output. This is highly dependent on hardware, but comparative benchmarks on standardized systems are essential.
- Benchmark Context: Runtime must be evaluated relative to dataset size (number of sequences) and complexity (sequence length, diversity). For iterative refinement in drug candidate screening, faster runtimes enable higher-throughput analysis.
Scalability
Scalability measures how runtime and memory usage increase as a function of input size (number of BCR sequences, n). It is the most critical KPI for applying methods to modern high-throughput B cell repertoire sequencing datasets.
- Assessment: Analyzed via complexity notation (e.g., O(n²), O(n³)) and empirical profiling. A method that scales poorly becomes computationally prohibitive for datasets exceeding a few thousand sequences.
- Bottlenecks: In BCR phylogenetics, the pairwise distance calculation and tree search space are common scalability challenges.
Experimental Protocol for KPI Benchmarking
A standardized protocol is required to fairly compare different phylogenetic tools (e.g., IgPhyML, dnaml, RAxML-NG, neighbor-joining implementations).
1. Dataset Simulation & Curation:
- Input: Use a known BCR lineage tree (e.g., from a well-characterized immune response or a simulated tree using tools like
DAWGorSIMPHY). - Process: Evolve BCR sequences along the branches of the known tree under a realistic SHM model (e.g., the GY94 codon model with hotspot targeting). This generates a sequence alignment with a known phylogenetic origin.
- Output: A "true tree" (Newick format) and the corresponding multiple sequence alignment (FASTA format).
2. Phylogenetic Inference:
- Apply each candidate tree inference method (Maximum Likelihood, Parsimony, Distance-based) to the same sequence alignment using default or optimized parameters for BCR data (accounting for SHM).
3. KPI Measurement:
- RF Distance: Compute RF distance between each inferred tree and the "true tree" using
Robinson-Fouldsfunctions in libraries likeDendropyorapein R. - Runtime: Log start and end times for each inference run. Perform multiple replicates to account for system noise.
- Scalability: Repeat steps 1-3 for datasets of increasing size (e.g., n=50, 100, 500, 1000, 5000 sequences). Plot runtime vs. n.
4. Data Aggregation & Analysis:
- Aggregate results across replicates and dataset sizes.
- Perform statistical tests (e.g., ANOVA) to determine if differences in KPIs between methods are significant.
The following tables summarize hypothetical but representative benchmark data from a recent comparative study of tools applicable to BCR phylogenetics.
Table 1: KPI Comparison Across Phylogenetic Methods (n=150 BCR Sequences)
| Method | Algorithm Class | Avg. RF Distance (±SD) | Avg. Runtime (seconds) | Memory Use (GB) |
|---|---|---|---|---|
| Tool A | Maximum Likelihood (SHM-optimized) | 0.08 (±0.02) | 285 | 1.2 |
| Tool B | Maximum Parsimony | 0.22 (±0.05) | 45 | 0.4 |
| Tool C | Neighbor-Joining (p-dist) | 0.19 (±0.04) | 12 | 0.1 |
| Tool D | Maximum Likelihood (general) | 0.10 (±0.03) | 620 | 2.5 |
Table 2: Scalability Profiling (Runtime in seconds)
| Number of Sequences (n) | Tool A (ML-SHM) | Tool B (Parsimony) | Tool C (NJ) | Tool D (ML-General) |
|---|---|---|---|---|
| 50 | 32 | 8 | <1 | 58 |
| 150 | 285 | 45 | 12 | 620 |
| 500 | 4,210 | 550 | 85 | 18,500 |
| 1000 | 15,800* | 2,100 | 310 | >48 hrs* |
*Extrapolated from model fit; indicates scalability limit.
Visualizing Methodologies and Relationships
Diagram 1: KPI Benchmarking Experimental Workflow
Diagram 2: Accuracy vs. Runtime Trade-off Space
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools & Reagents for BCR Phylogenetic Analysis
| Item | Function/Description | Example/Provider |
|---|---|---|
| BCR-Seq Library Prep Kit | Enriches and prepares BCR mRNA from B cell samples for high-throughput sequencing. Captures diversity. | SMARTer Human BCR Profiling (Takara Bio) |
| SHM-aware Codon Substitution Model | Evolutionary model for phylogenetic inference that accounts for the biased nature of SHM (e.g., hotspot targeting). | GY94 with SHM-specific parameters; implemented in IgPhyML. |
| Phylogenetic Inference Software | Core algorithm for building trees from aligned BCR sequences. Choice dictates KPIs. | IgPhyML (BCR-optimized ML), RAxML-NG, FastTree, PHYLIP. |
| Computational Benchmarking Suite | Scripts and pipelines to automate simulation, inference, and KPI calculation for fair comparison. | Custom Snakemake/Nextflow workflows; ETE Toolkit. |
| Tree Visualization & Annotation Tool | Enables biological interpretation of inferred trees (e.g., highlighting key mutations, clades). | ggtree (R), ITOL, Dendroscope. |
| "Ground Truth" Validation Set | Curated set of BCR sequences from in vitro or in vivo lineages with known relationships. Critical for validation. | Publicly available datasets from studies of well-defined immune responses (e.g., to influenza, HIV). |
For researchers investigating BCR somatic hypermutation patterns, rigorous evaluation of phylogenetic inference methods using the KPIs of RF Distance, Runtime, and Scalability is non-negotiable. The trade-offs are clear: BCR-optimized maximum likelihood methods typically offer the best accuracy (lowest RF distance) but at a higher computational cost. The choice of tool must be dictated by the specific research question, the scale of the dataset, and the required confidence in tree topology. As BCR repertoire sequencing scales towards millions of sequences, prioritizing scalable algorithms without completely sacrificing accuracy will be paramount for advancing vaccine and therapeutic antibody discovery.
This whitepaper provides an in-depth technical guide for integrating multi-omics data to elucidate the phylogenetic patterns of B cell receptor (BCR) somatic hypermutation (SHM). Within the broader thesis of BCR affinity maturation research, correlating phylogenetic trees derived from BCR sequencing with transcriptomic and proteomic profiles is critical for understanding the functional evolution of B cell clones during immune responses, with direct implications for vaccine design, autoimmune disease research, and therapeutic antibody development.
Foundational Concepts: BCR Phylogenetics and Multi-Omics
BCR SHM phylogenetics reconstructs the evolutionary history of a B cell clone from germline to its mutated progeny. Integrating this with other data layers allows researchers to map phenotypic changes onto phylogenetic branches.
Table 1: Core Multi-Omics Data Types in B Cell Research
| Data Type | Technology | Key Output | Relevance to BCR Phylogeny |
|---|---|---|---|
| BCR Repertoire Sequencing | Bulk/Single-cell V(D)J sequencing | Clonal lineages, mutation trees, SHM patterns | Provides the phylogenetic backbone (clonal trees). |
| Transcriptomics | RNA-seq (bulk or scRNA-seq) | Gene expression profiles, differential expression | Links branching events to changes in cell state, activation, or differentiation. |
| Proteomics | Mass spectrometry (LC-MS/MS), CyTOF | Protein abundance, post-translational modifications | Validates transcriptomic data and reveals functional protein-level adaptations. |
| Epigenomics | ATAC-seq, ChIP-seq | Chromatin accessibility, histone marks | Explains regulatory drivers of expression changes across lineages. |
Experimental Protocols for Integrated Data Generation
Protocol A: Coupled Single-Cell BCR and Transcriptome Sequencing
Objective: To generate paired BCR sequence and whole-transcriptome data from the same single B cell, enabling direct phylogenetic-transcriptomic correlation.
Detailed Methodology:
- Cell Preparation: Isolate B cells from lymphoid tissue or blood. Viability should be >90%.
- Single-Cell Partitioning: Load cells onto a platform like the 10x Genomics Chromium system using the 5' Gene Expression and V(D)J Immune Profiling solution.
- Library Preparation:
- GEM Generation & Barcoding: Cells are co-partitioned with gel beads in emulsion (GEMs). Within each GEM, reverse transcription occurs using barcoded primers.
- cDNA Amplification: Barcoded cDNA is PCR-amplified.
- Library Construction: The amplified cDNA is enzymatically fragmented and size-selected to construct the gene expression library. A separate, targeted PCR enriches V(D)J segments from the same cDNA pool for the BCR library.
- Sequencing: Pooled libraries are sequenced on an Illumina platform (e.g., NovaSeq). Recommended depth: ≥50,000 reads per cell for gene expression; ≥5,000 reads per cell for BCR.
- Data Processing: Use Cell Ranger (
cellranger multi) to align reads, quantify gene expression, and assemble contigs for BCR heavy and light chains.
Protocol B: Phylogenetic Tree Construction from BCR Sequences
Objective: To infer phylogenetic trees representing the SHM history of a B cell clone.
Detailed Methodology:
- Clonal Grouping: Group BCR sequences into clones using tools like
Change-OorScirpy. Criteria: same V and J gene segments, and a defined nucleotide distance threshold in the CDR3 region. - Multiple Sequence Alignment: For each clone, align the variable region sequences (from FR1 through FR3) using a tool like
MAFFTorClustal Omega. - Germline Reconstruction: Infer the unmutated germline ancestor sequence for the clone using
IgPhyMLor partis. - Phylogenetic Inference: Use
IgPhyML(which implements models of SHM) orFastTree/RAxML(with appropriate nucleotide models) on the aligned sequences, including the inferred germline as an outgroup root. - Tree Annotation: Annotate tree nodes with sequence features: mutation count, isotype (if known), and key amino acid changes.
Protocol C: Proteomic Profiling of Sorted B Cell Subsets
Objective: To quantify protein expression in B cell populations defined by phylogenetic position (e.g., early vs. late branches).
Detailed Methodology:
- Cell Sorting Based on Phylogeny: For a clone of interest, design FACS sorting panels using lineage markers (CD19, CD20) and clone-specific markers (unique BCR idiotype) or functional markers (e.g., CD27, CD38). Sort populations corresponding to key phylogenetic nodes.
- Sample Preparation: Lyse sorted cell pellets (min. 50,000 cells per sample) in RIPA buffer with protease inhibitors.
- Protein Digestion: Reduce with DTT, alkylate with iodoacetamide, and digest with trypsin/Lys-C overnight.
- LC-MS/MS Analysis: Desalt peptides and separate via nano-liquid chromatography coupled to a high-resolution tandem mass spectrometer (e.g., Q-Exactive HF).
- Data Analysis: Search MS/MS spectra against a human proteome database using software like MaxQuant or FragPipe. Use label-free quantification (LFQ) intensity for cross-sample comparison.
Data Integration and Correlation Strategies
The core challenge is mapping transcriptomic/proteomic data onto phylogenetic trees.
Strategy 1: Phenotype Mapping. Discrete cell states (e.g., naive, memory, plasmablast) from transcriptomics are mapped onto the tree tips, and ancestral state reconstruction is performed to infer transitions.
Strategy 2: Continuous Trait Correlation. Expression levels of key genes (e.g., MYC, BCL6, PRDM1) are treated as continuous traits. Tools like phytools in R can correlate trait evolution with branch lengths (mutation accumulation).
Table 2: Quantitative Correlation Example: SHM vs. Gene Expression
| Clone ID | Avg. SHM per Branch | AICDA Expression (TPM) |
BCL6 Expression (TPM) |
IRF4 Expression (TPM) |
Proteomic PTM Score (Activation) |
|---|---|---|---|---|---|
| Clone_001 | 12.4 | 45.2 | 32.1 | 5.1 | 0.85 |
| Clone_002 | 5.1 | 12.5 | 45.3 | 1.2 | 0.41 |
| Clone_003 | 18.7 | 67.8 | 18.9 | 25.4 | 0.92 |
| Clone_004 | 8.9 | 22.3 | 38.7 | 8.9 | 0.63 |
Note: TPM = Transcripts Per Million; PTM Score = Normalized phosphoprotein signal intensity related to B cell activation pathways.
Visualization of Integrated Workflows and Pathways
Title: Integrated BCR & Transcriptomic Single-Cell Workflow
Title: Signaling Linking Transcriptomics & SHM Phylogeny
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for Integrated BCR Multi-Omics
| Item | Function | Example Product/Catalog |
|---|---|---|
| Single-Cell 5' Immune Profiling Kit | Enables coupled V(D)J and gene expression profiling from the same single cell. | 10x Genomics, Chromium Next GEM Single Cell 5' Kit v3 |
| BCR/Germline-Specific Primers | For targeted amplification of BCR variable regions in bulk assays. | IGHV/IGKV/IGLV family-specific primer mixes (e.g., from Invitrogen). |
| B Cell Isolation/Culture Media | For maintaining B cell viability and specific differentiation states ex vivo. | IMDM + 10% FBS + human CD40L + IL-4 + IL-21 for plasma cell differentiation. |
| Antibody Panels for FACS | To sort B cell subsets or specific clones based on surface markers. | Anti-human CD19, CD20, CD27, CD38, IgD, plus anti-idiotype antibodies. |
| Phospho-Protein Antibodies (CyTOF) | For high-plex proteomic profiling of signaling states. | Maxpar Direct Immune Profiling Assay panels. |
| Cell Lysis Buffer for Proteomics | Efficient, MS-compatible protein extraction from low cell numbers. | RIPA Buffer supplemented with HALT Protease & Phosphatase Inhibitor Cocktail (Thermo). |
| Nucleotide Analogs (for Tracing) | To label dividing cells and link proliferation to phylogenetic branches. | 5-Ethynyl-2'-deoxyuridine (EdU). |
| Bioinformatics Pipelines | Essential software for data processing and integration. | Cell Ranger, IgPhyML, Change-O, Scirpy, Seurat, phytools R package. |
The analysis of B cell receptor (BCR) somatic hypermutation (SHM) phylogenetic patterns represents a paradigm shift in immuno-monitoring. Moving beyond simple clonality metrics, the detailed reconstruction of B cell lineage trees from high-throughput sequencing data captures the dynamics of affinity maturation. This technical guide frames these patterns within a broader thesis: that the topology, branch length, and selection pressure signatures of BCR phylogenies are non-invasive biomarkers predictive of clinical outcomes and therapeutic efficacy in oncology, autoimmune disorders, and infectious diseases.
Core Phylogenetic Signatures and Their Clinical Correlates
Quantitative metrics extracted from BCR lineage trees can be linked to distinct biological processes and patient states.
Table 1: Key Phylogenetic Signatures and Clinical Associations
| Signature Metric | Technical Definition | Proposed Biological Interpretation | Correlated Clinical Outcome (Examples) |
|---|---|---|---|
| Tree Balance (Colless Index) | Measures inequality of descendant leaves across internal nodes. | High imbalance suggests intense selection & clonal dominance. | Adverse in CLL; linked to aggressive disease & resistance. |
| Branch Length Skewness | Statistical skewness of path lengths from root to leaves. | Positive skew indicates a mix of naive and highly mutated cells. | Favorable in lupus; correlates with reduced renal flare risk. |
| Normalized Tree Diameter | Longest path between two leaves, divided by total mutations. | Measures diversification breadth vs. depth. | High in anti-PD-1 responders (melanoma). |
| Selection Pressure (dN/dS) | Ratio of non-synonymous to synonymous mutations in CDRs/FWRs. | >1 indicates antigen-driven selection. | Rising dN/dS post-vaccine correlates with neutralizing antibody titer. |
| Recent Expansion Index | Ratio of leaves within last 20% of tree depth to total leaves. | Quantifies recent clonal expansion. | High pre-treatment predicts rapid relapse in DLBCL. |
Detailed Experimental Protocol: From Sample to Phylogenetic Signature
Protocol 1: BCR Repertoire Sequencing & Lineage Tree Reconstruction
Objective: Generate high-fidelity BCR heavy-chain (IGH) sequences and reconstruct accurate phylogenetic lineages for a defined clonal family.
Materials & Workflow:
- Sample Input: PBMCs or tissue biopsy (fresh/frozen/FFPE).
- BCR Amplification: Use multiplex PCR primers (BIOMED-2 or equivalent) with unique molecular identifiers (UMIs). Critical: Sufficient input (≥10ng DNA) for low error rate.
- Sequencing: High-depth MiSeq (2x300bp) or NovaSeq for deeper repertoire.
- Bioinformatic Processing:
- Preprocessing & Clustering: UMI-aware error correction (e.g.,
pRESTO). Cluster sequences into clonal families based on V/J gene identity and CDR3 homology (Change-O). - Alignment & Tree Building: For each clone, perform multiple sequence alignment (
Clustal Omega). Reconstruct maximum-likelihood phylogenetic tree (IgPhyML- models SHM kinetics) or neighbor-joining tree (fasta-based tools). - Signature Extraction: Calculate metrics in Table 1 using
adephylo(R) or custom Python scripts (ETE3 toolkit).
- Preprocessing & Clustering: UMI-aware error correction (e.g.,
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| UMI-linked BCR Amplification Kit (e.g., Takara Bio SMARTer Human BCR Kit) | Reduces PCR and sequencing errors, enabling true lineage variant calling. |
| Spike-in Synthetic BCR Standards | Quantifies absolute clonal frequency and controls for amplification bias. |
| IgPhyML Software | Phylogenetic inference tool specifically designed for BCR sequences incorporating SHM models. |
PhyDyn Suite (or BEAST2) |
For more advanced, time-scaled phylogenetic analysis to estimate growth rates. |
| Validated Anti-IgG/A/M Capture Beads | For isotype-specific BCR repertoire analysis, linking phylogeny to function. |
Validation Workflow: Correlating Signatures with Outcomes
Protocol 2: Longitudinal Tracking of Phylogenetic Metrics
Objective: Statistically link temporal changes in phylogenetic signatures to therapeutic response.
- Cohort Design: Prospectively collect serial blood/tissue samples at baseline (T0), on-treatment (T1), and progression/relapse (T2).
- Data Acquisition: Process all samples identically per Protocol 1.
- Feature Matrix: For each patient timepoint, generate a matrix of the phylogenetic metrics (Table 1) for their top 10-100 clones by frequency.
- Statistical Modeling:
- Unsupervised: Hierarchical clustering of patients based on phylogenetic feature matrix.
- Supervised: Train a Cox proportional-hazards model using baseline signatures to predict PFS/OS. Use mixed-effects models to test if specific metric trajectories differ between responders/non-responders.
Diagram 1: Clinical Validation Workflow (77 chars)
Mechanistic Links: Signaling Pathways Implicated in Phylogenetic Outcomes
Phylogenetic patterns reflect underlying B cell biology. Key pathways influencing these signatures include:
- B Cell Receptor (BCR) Signaling: Chronic antigen engagement drives sustained SHM and selection (high dN/dS).
- T Cell Help (CD40L/CD40, ICOS): Essential for germinal center formation and proper affinity maturation; its absence leads to aberrant tree topologies.
- Cytokine Signals (IL-21, IL-4): Shape the mutational landscape and isotype switch, affecting branch lengths.
Diagram 2: B Cell Signaling & Phylogenetic Outcomes (71 chars)
Applications in Drug Development
Table 2: Utility of Phylogenetic Signatures in Therapeutic Contexts
| Development Stage | Application | Example |
|---|---|---|
| Target Discovery | Identify pathogenic clones driving autoimmunity via unique tree shapes. | Trees with extreme imbalance in RA synovium. |
| Patient Stratification | Enrich trials with patients likely to respond based on baseline signatures. | High normalized diameter for immunotherapy trials. |
| Pharmacodynamics | Early biomarker of target engagement (changes in selection pressure). | dN/dS shift after BTK inhibitor in CLL. |
| Resistance Monitoring | Detect outgrowth of minor, treatment-resistant subclones early. | Emergence of new, short-branch trees at progression. |
The clinical validation of BCR phylogenetic signatures requires standardized wet-lab protocols, robust bioinformatic pipelines, and rigorous statistical correlation in well-annotated cohorts. The frontier lies in integrating these B cell lineage trees with T cell receptor phylogenies and tumor genomic data to build a complete picture of adaptive immune pressure. This approach promises to deliver powerful, dynamic biomarkers for personalized medicine.
Conclusion
The phylogenetic analysis of B cell receptor somatic hypermutation patterns has evolved from a conceptual model into a powerful, quantitative framework central to modern immunology and translational medicine. By understanding the foundational evolutionary principles (Intent 1), researchers can effectively apply sophisticated computational pipelines to trace antibody lineages in health and disease (Intent 2). Success requires navigating analytical pitfalls with optimized, best-practice protocols (Intent 3), rigorously validated by standardized comparative metrics (Intent 4). Looking forward, the integration of BCR phylogenetics with single-cell multi-omics and spatial transcriptomics promises unprecedented resolution of the germinal center reaction. This will accelerate the discovery of diagnostic biomarkers, forecast vaccine efficacy, and guide the rational design of monoclonal antibodies and B cell-targeted therapies, ultimately bridging deep immunological insight with tangible clinical impact.