Unlocking Rare Clones: A Practical Guide to Optimizing MiXCR's Low-Percentage Read Analysis for Immunome Research
This article provides a comprehensive framework for handling low-percentage sequencing reads in MiXCR clonotype analysis.
Unlocking Rare Clones: A Practical Guide to Optimizing MiXCR's Low-Percentage Read Analysis for Immunome Research
Abstract
This article provides a comprehensive framework for handling low-percentage sequencing reads in MiXCR clonotype analysis. Targeting researchers and drug development professionals, it covers the foundational biology of rare clones, methodological best practices for accurate assembly, troubleshooting common pitfalls like false positives and PCR errors, and validation strategies against other tools like IgBLAST and VDJtools. The goal is to empower users to confidently extract meaningful biological signals from low-abundance data critical for minimal residual disease (MRD) detection, vaccine response monitoring, and autoimmune disorder research.
Why Rare Clones Matter: The Biological Significance of Low-Abundance Reads in Immune Repertoire Sequencing
Troubleshooting Guides & FAQs
Q1: During MiXCR analysis, my final clonotype table contains many clones with very low percentages (<0.01%). Are these real T-cell/B-cell clones or just background noise from PCR/sequencing errors? A: Low-frequency clones (<0.01%) can represent true, rare clonotypes but are often near the technical noise floor. Key factors to check:
- UMI Deduplication: Ensure Unique Molecular Identifiers (UMIs) were used and correctly processed during the
alignandassemblesteps to collapse PCR duplicates. - Error Correction: Verify that the
assemblestep's--error-correctionparameters are optimized for your data type (e.g.,--error-correction bwafor high-quality data). - Spike-in Controls: Use synthetic immune receptor spike-ins at known, low concentrations to empirically define the detection limit for your specific wet-lab and analysis protocol.
Q2: How do I determine the appropriate minimum clone count or percentage threshold for downstream analysis (e.g., tracking clones over time, identifying responders in drug trials)? A: There is no universal threshold. You must establish it empirically based on:
- Negative Controls: Process negative control samples (no template, healthy donor not exposed to antigen) through the same pipeline. The maximum clonal size observed in these controls defines a conservative "background" threshold.
- Sequencing Depth: The threshold is relative to your total sequencing reads. A deeper sequenced sample will reliably detect lower percentages. Calculate the theoretical detection limit as (1 / total productive reads) * 100.
Q3: I am comparing pre- and post-treatment samples. A key therapeutic clone appears to drop from 5% to 0.1%. Is this a biologically significant decrease or an artifact of differing sample cellularity/sequencing depth? A: Normalize your data. Do not rely on raw percentages.
- Input Normalization: Use an equal number of input cells for library prep.
- Downsampling: In your analysis, use the
--downsamplingoption in MiXCR'sexportcommand to normalize all samples to the same number of sequencing reads before calculating frequencies. This allows for direct comparison.
Q4: When using the clonotypes fix command in MiXCR, what do the parameters --low-percentage-bound and --high-percentage-bound actually do, and how should I set them?
A: The clonotypes fix command is used to correct for cross-contamination between samples in a multiplexed run. It identifies and removes "floating" clones.
- Principle: A true, biologically expanded clone should be present at a stable, detectable frequency in its sample of origin and be absent (or at a very low, constant background level) in all other samples.
- Parameter Function:
--low-percentage-bound: Sets the maximum frequency at which a clone is considered "absent" or background in a sample where it is not the major sample. Clones below this threshold in non-major samples are considered cross-contamination and can be subtracted. (Typical setting: 0.001% to 0.01%).--high-percentage-bound: Sets the minimum frequency for a clone to be considered "present" in its major sample. (Typical setting: 0.1% to 0.5%).
- Setting Guidelines: These must be determined from your negative control samples and the observed level of index hopping/cross-talk in your sequencing run.
Experimental Protocols
Protocol 1: Empirical Determination of Detection Sensitivity using Spike-ins
- Spike-in Reagent Preparation: Dilute a commercially available synthetic T-cell/B-cell receptor reference standard (e.g., from Horizon Discovery) to create a dilution series spanning from 0.0001% to 1% in a background of carrier RNA.
- Library Preparation & Sequencing: Process each spike-in dilution alongside your experimental samples using the identical wet-lab protocol (RNA extraction, cDNA synthesis, UMI-based immune repertoire library kit, sequencing).
- MiXCR Analysis: Process all samples with a standardized MiXCR pipeline (e.g.,
mixcr analyze shotgun ...). - Data Analysis: For each spike-in clone, plot its input percentage against its detected percentage. The point where the curve deviates from linearity defines your assay's lower limit of quantification (LLOQ). The lowest consistently detected percentage defines the limit of detection (LOD).
Protocol 2: Establishing Background Threshold with Negative Controls
- Sample Preparation: Include at least three types of negative controls in every sequencing batch:
- No Template Control (NTC): Water instead of RNA/cDNA.
- Negative Biological Control: RNA from a cell line lacking immune receptors (e.g., HEK293).
- Pooled Healthy Donor Control: To establish a polyclonal baseline.
- Analysis: Run all controls through your MiXCR pipeline. For the NTC and cell line controls, any clonotype called is technical noise. The 99th percentile of clone frequencies in the polyclonal healthy donor can serve as a biological "non-expanded" baseline.
- Threshold Setting: Set your
--low-percentage-boundforclonotypes fixabove the maximum frequency observed in the NTC.
Data Presentation
Table 1: Impact of Sequencing Depth on Low-Percentage Clone Detection
| Total Productive Reads | Theoretical Minimum Detectable % (1/Reads)*100 | Empirically Determined LOD (via Spike-in) | Recommended Conservative Threshold for Analysis |
|---|---|---|---|
| 50,000 | 0.002% | 0.005% | 0.01% |
| 100,000 | 0.001% | 0.002% | 0.005% |
| 500,000 | 0.0002% | 0.0008% | 0.001% |
| 1,000,000 | 0.0001% | 0.0005% | 0.001% |
Table 2: clonotypes fix Parameter Scenarios & Outcomes
| Scenario | --low-percentage-bound |
--high-percentage-bound |
Effect on Clonal Data |
|---|---|---|---|
| Too Stringent | 0.0001% | 1.0% | Over-correction: May remove true, low-frequency shared clones (e.g., public clones). |
| Too Permissive | 0.1% | 0.01% | Under-correction: Fails to remove substantial cross-contamination, leaving artifactual clones. |
| Balanced (Example) | 0.005% | 0.1% | Optimal: Removes index-hopping artifacts while preserving true, low-frequency biology. Assumes LOD of ~0.001%. |
Visualizations
Title: MiXCR Clonotype Fix Workflow with Thresholds
Title: Clonotype Fix Logic: Removing Cross-Contamination
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Low-% Clonotype Research |
|---|---|
| UMI-based Immune Repertoire Library Prep Kit (e.g., from Takara Bio, BioLegend) | Contains unique molecular identifiers (UMIs) to tag original mRNA molecules, enabling precise removal of PCR duplicates and error correction to reveal true low-frequency clones. |
| Synthetic Immune Receptor Spike-in Controls (e.g., from Horizon Discovery) | Artificially engineered clones at known, low frequencies. Used to empirically determine the Limit of Detection (LOD) and Limit of Quantification (LOQ) of the entire workflow. |
| High-Fidelity DNA Polymerase (e.g., Q5, KAPA HiFi) | Minimizes PCR amplification errors during library construction, reducing sequencing noise that can be misidentified as low-frequency clonotypes. |
| Cell Sorting Reagents (Antibodies, Beads) | For pre-enriching specific lymphocyte populations (e.g., CD8+ T-cells), increasing the relative frequency of antigen-specific clones and improving the signal-to-noise ratio for detection. |
| Multiplexing Indexes with Unique Dual Indexes (UDI) | Minimizes index hopping (crosstalk) between samples during sequencing, a major source of artificial low-frequency "contaminant" clones that must be filtered by clonotypes fix. |
Technical Support Center: MiXCR Low-Percentage Clonotype Analysis
Troubleshooting Guides & FAQs
Q1: My MiXCR analysis reports clonotypes with very low read percentages (<0.01%). Are these real biological signals or technical noise? A: Rare clones with low percentages can be biologically relevant, especially in early disease or minimal residual disease (MRD) contexts. To validate:
- Check sequencing depth: Ensure sufficient input reads (≥ 100,000 for TCR/IG). Low depth inflates rare clone noise.
- Replicate the experiment: A true biological signal should appear, albeit at low frequency, in independent library preparations from the same sample.
- Apply a confidence threshold: Use a clone-specific quality threshold (e.g.,
-OvParameters.parameters.qualityThreshold=20inmixcr analyze). - Examine UMIs/UMI groups: If using UMI-based protocols, require ≥2 reads per UMI group to call a clone, filtering PCR errors.
Q2: How can I optimize MiXCR parameters to improve sensitivity for rare, low-percentage clones without capturing excessive artifacts?
A: Adjust the align and assemble steps. A recommended workflow for rare clone detection:
Q3: When tracking rare clones across serial samples (e.g., pre- and post-treatment), what is the best method to ensure consistent identification?
A: Use MiXCR's assembleContigs with the same reference set. Process all samples separately through align and assemblePartial. Then, pool .clns files for joint assembly:
This ensures consistent CDR3 clustering and V/J gene assignment across the dataset, critical for reliable rare clone tracking.
Q4: How should I handle low-frequency clones in downstream statistical analysis (e.g., diversity metrics, differential abundance)? A: Low-frequency clones require specialized statistical approaches to avoid bias.
- For Diversity Indices: Use metrics that account for sampling depth and clone distribution, such as the normalized Shannon diversity index or Hill numbers. Avoid relying solely on the raw clone count.
- For Differential Abundance: Employ methods designed for sparse compositional data, like ALDEx2 or ANCOM-BC, which account for the compositional nature of repertoire data and low counts.
Key Quantitative Data in Rare Clone Analysis Table 1: Typical Thresholds and Their Implications
| Parameter | Typical Value | Purpose | Risk if Too Low | Risk if Too High |
|---|---|---|---|---|
| Minimal Read Count | 2-5 reads | Filter PCR/sequencing errors | High false positives (noise) | Loss of true rare clones |
| Minimal Percentage | 0.0001 - 0.001% | Context-dependent biological filter | Includes noise | Misses clinically relevant MRD |
| Sequencing Depth | 100,000 - 1M+ reads | Sufficient sampling of repertoire | Poor clone quantification | Diminishing returns on cost |
| UMI/Min Group Size | 2-3 reads/group | Error correction for UMI protocols | Ineffective error correction | Loss of low-UMI-count clones |
Detailed Experimental Protocol: Rare Clone Detection and Validation
Title: Longitudinal Tracking of Rare Clonotypes in Therapeutic Response Monitoring.
Objective: To identify and validate rare, disease-relevant T-cell or B-cell clones present at low frequencies in serial patient samples.
Materials: See "Research Reagent Solutions" table below.
Methodology:
- Sample Preparation: Isolate PBMCs or tissue-derived lymphocytes at multiple time points (e.g., baseline, on-treatment, relapse). Extract high-quality total RNA/DNA.
- Library Construction: Use a UMI-based multiplex PCR protocol (e.g., SMARTer Human TCR a/b Profiling Kit) for accurate error correction and molecule counting. Perform technical replicates.
- High-Throughput Sequencing: Sequence on a platform allowing sufficient depth (e.g., Illumina MiSeq, 2x300 bp). Aim for ≥500,000 raw read pairs per sample.
- Bioinformatic Processing with MiXCR:
- Run the standardized
mixcr analyzepipeline with theumipreset, adjusting the--minimal-clone-countparameter to 2. - Export the clone table for all samples with a very low fractional threshold (
fractionOfTotal >= 1e-6).
- Run the standardized
- Cross-Sample Analysis: Use the
assembleContigsmethod (described in FAQ A3) to generate a consistent clonotype set across all samples. - Validation by Targeted PCR: For top candidate rare clones, design clone-specific primers for the CDR3 region. Perform nested digital droplet PCR (ddPCR) on original cDNA to obtain an absolute, amplification-bias-free quantification.
- Statistical & Biological Correlation: Correlate the dynamics of validated rare clones with clinical parameters (e.g., tumor burden, drug response) using longitudinal models.
Diagrams
Title: Rare Clone Analysis Workflow
Title: Signal vs. Noise Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Rare Clonotype Research
| Item | Function in Rare Clone Analysis |
|---|---|
| UMI-based TCR/IG Profiling Kit(e.g., SMARTer Human TCR a/b) | Adds Unique Molecular Identifiers (UMIs) to each starting molecule during cDNA synthesis, enabling precise error correction and absolute molecule counting. Critical for distinguishing true rare clones from PCR/sequencing errors. |
| High-Fidelity PCR Enzyme Mix | Minimizes PCR amplification bias and errors during library amplification, ensuring more accurate representation of initial clone frequencies. |
| ddPCR Supermix for Probes | Enables absolute quantification of specific CDR3 sequences (clonotypes) without reliance on standards, providing gold-standard validation for rare clones identified by NGS. |
| Clonotype-Specific Primers/Probes | Custom-designed TaqMan assays for the CDR3 region of a specific clone. Used in ddPCR validation to confirm NGS findings and track clones with maximum sensitivity. |
| MiXCR Software | The core bioinformatic tool for aligning, assembling, and quantifying immune receptor sequences from raw NGS data. Its parameters are tuned for sensitive rare clone detection. |
| Spike-in Synthetic TCR/IG Standards | Known, low-abundance sequences added to the sample pre-processing. Used to calibrate sensitivity, quantify detection limits, and monitor technical performance across runs. |
Technical Support Center: Troubleshooting MiXCR for Low Percentage Clonotype Analysis
This support center addresses common challenges researchers face when using MiXCR to identify and validate low-abundance (<0.01%) clonotypes, a critical task for the thesis on "Advancing Clonotype Resolution: Strategies for Reliable Detection of Low-Frequency Sequences in MRD and Immune Repertoire Profiling".
FAQ & Troubleshooting Guides
Q1: My MiXCR analysis reports clonotypes at ~0.1%, but spike-in controls suggest sensitivity should be 0.01%. What are the primary causes? A: The discrepancy often stems from pre-analytical or analytical bottlenecks.
- Cause 1: PCR Duplication Bias. Limited input DNA/RNA leads to stochastic amplification, skewing clonotype representation.
- Cause 2: Sequencing Error Noise. Errors in low-quality read regions are misassembled into artificial, low-frequency clonotypes.
- Cause 3: Inefficient Hybrid Capture (for DNA panels). Poor probe design or hybridization conditions can cause on-target inefficiency, dropping some clones below detection.
Mitigation Protocol:
- Input Material: Use >100ng of high-quality input gDNA or >50ng of total RNA. For single-cell protocols, ensure cell viability >90%.
- UMI Integration: Use a UMI-based (Unique Molecular Identifier) library prep kit. During MiXCR analysis, enforce the
--use-umisflag inmixcr analyzepipeline. - Error Correction: Apply stringent quality trimming (
--quality-trimming q30). In theassemblestep, use--error-correct cost-umisto correct errors based on UMI groups.
Q2: How can I distinguish true low-frequency clonotypes from technical artifacts or sequencing errors? A: Implement a multi-step validation filter. The following table summarizes quantitative thresholds derived from recent benchmarking studies:
| Filter Parameter | Recommended Threshold | Purpose |
|---|---|---|
| Reads per UMI Group | ≥ 3 | Confirms consistent PCR amplification of the original molecule. |
| UMI Count per Clonotype | ≥ 2 | Requires independent capture of at least two original molecules. |
| Clonal Quality Score (MiXCR) | ≥ 50 | Ensures high-confidence V/J alignment and assembly. |
| Background Error Rate | < 0.001% | Establish from negative control (no template) runs. |
Validation Protocol:
- Run Negative Controls: Include a no-template control (NTC) and a polyclonal healthy donor sample in every batch.
- Apply Filters: Post-MiXCR, use
mixcr exportClonesand filter clones against the table above using R/Python. - Wet-Lab Validation: For critical clones (e.g., MRD targets), design clone-specific qPCR assays or use targeted deep sequencing for confirmation.
Q3: When comparing time points for MRD, what is the optimal way to track low-percentage clonotypes while accounting for repertoire shifts? A: Use a normalized tracking method focused on clonal abundance relative to total productive reads.
Workflow for Longitudinal MRD Tracking:
- Standardized Preprocessing: Process all samples through the same MiXCR pipeline with identical parameters (
mixcr analyze --starting-material rna --species hs --only-productive). - Clone Alignment: Use the
mixcr overlayClonesfunction to create a unified table of clonotypes across all time points. - Abundance Normalization: For each clone at each time point, calculate:
(Clone Count / Total Productive Reads) * 100,000. - Apply Detection Limit: Set a positivity threshold based on your negative control's background (e.g., mean + 5 SD of background "clones").
Diagram: Workflow for Low-Abundance Clonotype Validation
Diagram: Key Decision Points in Low-Frequency Analysis
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Low-% Clonotype Research |
|---|---|
| UMI-Compatible cDNA Synthesis Kit (e.g., SMARTer TCR/BCR) | Adds unique molecular identifiers during reverse transcription to tag each original mRNA molecule, enabling error correction and digital counting. |
| Hybrid-Capture BCR/TCR Panels | Target enrichment panels for DNA-based MRD; ensure high uniformity and on-target rate to evenly capture all clonotypes. |
| Spike-in Synthetic Clonotype Controls | Artificially synthesized TCR/BCR sequences at known low abundances (e.g., 0.001%, 0.01%) to empirically define assay sensitivity and LOD. |
| High-Fidelity PCR Master Mix | Polymerase with ultra-low error rates is critical for minimizing mutations during library amplification that create false low-frequency variants. |
| qPCR Assay for Clone-Specific Validation | TaqMan assays designed against the CDR3 region of a clonotype of interest for orthogonal, sensitive validation outside NGS. |
Technical Support Center: Troubleshooting Low Clonotype Recovery in MiXCR
Frequently Asked Questions (FAQs)
Q1: After running MiXCR, my final clonotypes consist of a very low percentage (e.g., <5%) of the input sequencing reads. What are the primary causes? A: This is a common signal-vs-noise challenge. Primary causes include: 1) Excessive PCR/sequencing errors generating artificial diversity, 2) Insufficient or low-quality starting material leading to high technical noise, 3) Overly stringent alignment parameters discarding true but low-quality signals, 4) Failure to merge paired-end reads correctly, causing read drop-off, and 5) High levels of non-T/B cell reads in the sample (e.g., from poor cell sorting or RNA contamination).
Q2: How can I determine if my low clonotype recovery is due to biological reality (few true clones) or technical artifacts? A: Incorporate a spike-in control of known synthetic T-cell or B-cell receptor sequences at defined abundances. Analyze the control's recovery rate. If control recovery is high, the issue is likely biological/experimental. If control recovery is also low, the issue is technical (e.g., library prep, sequencing, or analysis parameters).
Q3: What specific MiXCR parameters should I adjust to improve sensitivity without drastically increasing false positives?
A: A staged approach is recommended. First, ensure you are using the latest version of MiXCR. Key parameters to adjust in the align and assemble steps include:
--report: Always generate and review the alignment report.- Alignment
--parameters: Consider relaxing--max-hits-to-tryand--max-hits-to-selectfor complex repertoires. assemble--minimal-scoreand--minimal-sum-qual: Lower these thresholds incrementally, but monitor the increase in clonotypes with single-read support.assembleContigs(for RNA-seq): Crucial for reconstructing full-length sequences from fragmented data.
Q4: My negative control (no template, healthy tissue) shows numerous clonotypes. How do I filter these out? A: Background noise is a critical confounder. Implement experimental and computational filtration:
- Experimental: Use unique molecular identifiers (UMIs) during library preparation to correct for PCR duplicates and errors.
- Computational: Use MiXCR's
exportCloneswith--filter "isFunctional"to keep only productive sequences. Subsequently, use the--specific-tagoption if you used sample multiplexing. Finally, perform cross-sample comparison: subtract clonotypes present in your negative control(s) that are below a defined frequency threshold (e.g., 0.1%) in your experimental sample.
Troubleshooting Guides
Issue: High Proportion of Reads Discarded in Alignment Stage
Symptoms: The align report shows a low "Successfully aligned reads" percentage.
Steps:
- Check Read Quality: Run FastQC on your raw FASTQ files. Look for poor base quality scores, adapter contamination, or abnormal nucleotide composition.
- Verify Species and Loci: Ensure the
--speciesand--loci(e.g.,TRB,IGH) parameters are set correctly in themixcr aligncommand. - Adjust Alignment Rigor: If using default parameters, try the
mixcr alignpresets for your data type:- For amplicon data:
--preset amplicon - For RNA-seq data:
--preset rna-seq
- For amplicon data:
- Check for Contamination: Align a subset of unaligned reads to the host genome (e.g., human) using a tool like Bowtie2. A high alignment rate may indicate genomic contamination drowning out TCR/IG signals.
Issue: Many Singleton Clonotypes (Clonotypes supported by only one read) After Assembly
Symptoms: The assemble report or exported clonotype table shows most clonotypes have a "Read count" of 1.
Steps:
- Enable UMI Processing: If your data contains UMIs, use the
--umi-positionand related parameters inmixcr preprocessandmixcr assemble --use-umisto collapse PCR duplicates and errors. - Increase Clustering Thresholds: In
mixcr assemble, increase the--minimal-clonal-occupancy(e.g., from 2 to 3) to require more overlapping reads to form a clonotype. Also review--cluster-for-identity. - Review Error Correction: The
mixcr assemblestep includes error correction. If--minimal-sum-qualis too high, true low-quality reads from a rare clone may not be error-corrected and merged. If it's too low, noise will cluster. Adjust based on your sequencing quality. - Post-hoc Filtering: After exporting clonotypes, filter your final list to exclude singletons or those below a frequency threshold (e.g., 0.01%). Use this with caution, as it may remove ultra-rare but true signals.
Experimental Protocols for Validating Low-Abundance Clonotypes
Protocol 1: Spike-in Control Validation for Sensitivity Assessment Objective: Quantify the technical sensitivity limit of your MiXCR workflow. Materials: See "Research Reagent Solutions" table. Method:
- Spike-in Addition: Prior to RNA extraction or library amplification, add 1 µL of the ART-Spike Mix to your sample. Record the exact number of molecules added for each synthetic clonotype.
- Proceed with Standard Workflow: Perform RNA extraction, cDNA synthesis (using a gene-specific primer for the constant region if possible), library preparation, and sequencing.
- Dedicated Analysis: Run MiXCR analysis on the sequenced sample with your standard parameters.
- Recovery Calculation: In the output clonotypes, identify the spike-in sequences by their CDR3 amino acid sequence. Calculate recovery as: (Observed spike-in read count) / (Expected spike-in read count based on added molecules and sequencing depth) * 100%.
Protocol 2: Biological Replicate Concordance Check Objective: Distinguish stochastic noise from reproducible, low-abundance biological signal. Method:
- Process Replicates Independently: Starting from biological material (e.g., split cell aliquots), carry out the entire wet-lab and computational workflow independently for at least 3 replicates.
- Comparative Analysis: Use MiXCR's
mixcr overlapcommand to calculate the overlap coefficient between the clonotype sets of each replicate pair. - Signal Identification: Clonotypes that appear in 2/3 or 3/3 replicates are highly likely to be true biological signals, even if their frequency is low in each individual run. Clonotypes appearing in only one replicate are likely technical noise or stochastic transcriptional events.
Data Presentation
Table 1: Impact of Key MiXCR assemble Parameters on Clonotype Recovery and Noise
| Parameter | Default Value | Recommended Range for Low-Abundance | Effect of Increasing Value | Risk of Lowering Value |
|---|---|---|---|---|
--minimal-score |
50 | 30 - 50 | Reduces sensitivity, may drop true rare clones. | Increases noise (more false clonotypes). |
--minimal-sum-qual |
50 | 40 - 60 | Similar to above, filters based on Phred quality. | Similar to above. |
--minimal-clonal-occupancy |
2 | 2 - 3 | Aggressively removes singleton noise. | Increases singletons, potential false positives. |
--cluster-for-identity (CDR3 AA) |
1.0 | 0.9 - 1.0 | Merges highly similar sequences (error correction). | Over-clusters, losing true diversity. |
Table 2: Typical Read Fate in MiXCR Analysis of Human TRB Repertoire from RNA-seq
| Processing Stage | Percentage of Input Reads (Typical Range) | Common Reason for Read Loss |
|---|---|---|
| Initial Input Reads | 100% | - |
After Alignment (align) |
60% - 85% | Low quality, non-TRB origin, adapter dimers. |
After Assembly & Error Correction (assemble) |
40% - 70% of aligned | Filtering by quality, failure to cluster. |
| In Final Exported Functional Clonotypes | 20% - 60% of assembled | Non-productive sequences, in-frame stops. |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Context of MiXCR/Clonotyping |
|---|---|
| UMI-Adopted Library Prep Kits (e.g., SMARTer TCR a/b Profiling Kit) | Integrates Unique Molecular Identifiers (UMIs) during cDNA synthesis to tag original molecules, enabling precise correction for PCR and sequencing errors, critical for accurate quantification of rare clones. |
| Synthetic Spike-in Control Libraries (e.g., ART-Spike) | Contains known, non-natural TCR/BCR sequences at defined ratios. Added to samples pre-processing to benchmark sensitivity, quantify technical dropout, and normalize between runs. |
| Magnetic Cell Separation Kits (e.g., Pan T Cell Isolation Kit) | Enriches target lymphocyte population (T/B cells) from PBMC or tissue, increasing the fraction of relevant signal reads and reducing noise from other cell types. |
| High-Fidelity PCR Enzymes (e.g., Q5, KAPA HiFi) | Minimizes PCR-induced errors during target amplification and library construction, reducing artificial diversity that is misinterpreted as noise or rare clonotypes. |
| Dual-Indexed Sequencing Adapters | Allows high-level multiplexing of samples while reducing index hopping (barcode swapping), which can create cross-sample contamination noise. |
Visualizations
Diagram 1: MiXCR Workflow for Maximizing Signal Recovery
Diagram 2: Sources of Noise vs. Signal in Immune Repertoire Sequencing
Step-by-Step: Best Practices for MiXCR Assembly and Reporting of Low-Frequency Clonotypes
Troubleshooting Guides & FAQs
Q1: After running MiXCR, my clonotype table is dominated by very low percentage reads (<0.1%). What is the first pre-processing step I should check?
A: Immediately verify the completeness of Adapter Trimming. Residual adapter sequences cause misalignment of reads, generating a high number of spurious, low-count clonotypes. Use FastQC on your raw FASTQ files and look for overrepresented sequences in the "Adapter Content" plot. Re-run trimming with a tool like cutadapt, explicitly specifying your library preparation kit's adapter sequences.
Q2: My sequencing run has overall good quality scores, but MiXCR still reports many low-frequency clonotypes. Could underlying errors be the cause?
A: Yes. Even with Phred scores >Q30, cumulative PCR and sequencing errors can create artificial diversity. Implement an Error Correction step before MiXCR alignment. Use a tool like bfc, Racer, or HiFi-mode in cutadapt for overlapping paired-end reads. This step collapses error-containing reads into a single, high-quality consensus read, dramatically reducing noise.
Q3: What are the critical Quality Control (QC) metrics post-preprocessing that specifically impact low-abundance clonotype detection in MiXCR? A: Focus on three post-trimming/cleaning metrics. Failures here directly increase background noise.
Table 1: Critical Post-Preprocessing QC Metrics for MiXCR Sensitivity
| Metric | Target Value | Impact on Low-% Clonotypes |
|---|---|---|
| % Adapter Content | 0% | High levels create false alignments and spurious clonotypes. |
| % of Reads Retained | >85% (post-trim) | Aggressive trimming can remove true CDR3 regions, skewing repertoire. |
| Mean Read Length | Consistent with expected amplicon length | Shortened reads may lack V/J primer regions, causing MiXCR to discard them. |
Q4: I've performed trimming and error correction. Which MiXCR parameters should I adjust next to handle residual low-frequency noise?
A: Tune the --downsampling and --error-correcting parameters within MiXCR. Use --downsampling none to preserve all quantitative data for frequency analysis. Aggressively set the --error-correcting parameters (--error-correcting align,assemble) to group together reads originating from the same clonotype but containing minor errors.
Q5: How do I design a workflow that integrates all these steps for optimal sensitivity in clonotype fixation studies? A: Follow a sequential, QC-gated pipeline. The diagram below outlines the critical path.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for High-Sensitivity TCR/BCR Repertoire Sequencing
| Item | Function in Pre-Processing Context |
|---|---|
| Strand-Specific Adapter Kit (e.g., Illumina TruSeq) | Defines the exact adapter sequences for trimming; critical for accurate cutadapt command. |
| UMI (Unique Molecular Identifier) Oligos | Integrated into library prep; allows for precise error correction and PCR duplicate removal before alignment, the gold standard for noise reduction. |
| High-Fidelity PCR Polymerase (e.g., Q5, KAPA HiFi) | Minimizes PCR errors introduced during library amplification, reducing the burden on subsequent error-correction steps. |
| Size Selection Beads (e.g., SPRIselect) | Ensures removal of primer-dimers and overly short fragments that consume sequencing depth and contribute to low-quality alignments. |
Dedicated Trimming Software (cutadapt, fastp, Trimmomatic) |
Performs the precise removal of non-biological sequence, preventing misalignment in MiXCR. |
Detailed Protocol: Integrated Pre-Processing for MiXCR
Objective: Generate clean, error-corrected FASTQ files from raw immune repertoire sequencing data to maximize true clonotype detection sensitivity in MiXCR.
Materials:
- Raw paired-end FASTQ files (R1, R2).
- Adapter sequences (e.g.,
AGATCGGAAGAGCfor Illumina). cutadapt(v4.0+),FastQC(v0.11.9+),bfc(v181) orRACER(v1.1.1),MiXCR(v4.0.0+).
Methodology:
Initial Quality Assessment:
Inspect
fastqc_raw/*/htmlreports. Note adapter overrepresentation and sequence length.Adapter and Quality Trimming:
Replace
ADAPTER_FWD/ADAPTER_REVwith your specific sequences.Post-Trim QC:
Confirm near-zero adapter content and assess read loss.
Error Correction & Deduplication (using RACER):
This merges paired ends, corrects errors, and outputs a single, high-quality consensus FASTQ.
Final Data Preparation for MiXCR:
The
--adapters no-adaptersand--error-correctingflags are now valid due to prior pre-processing.
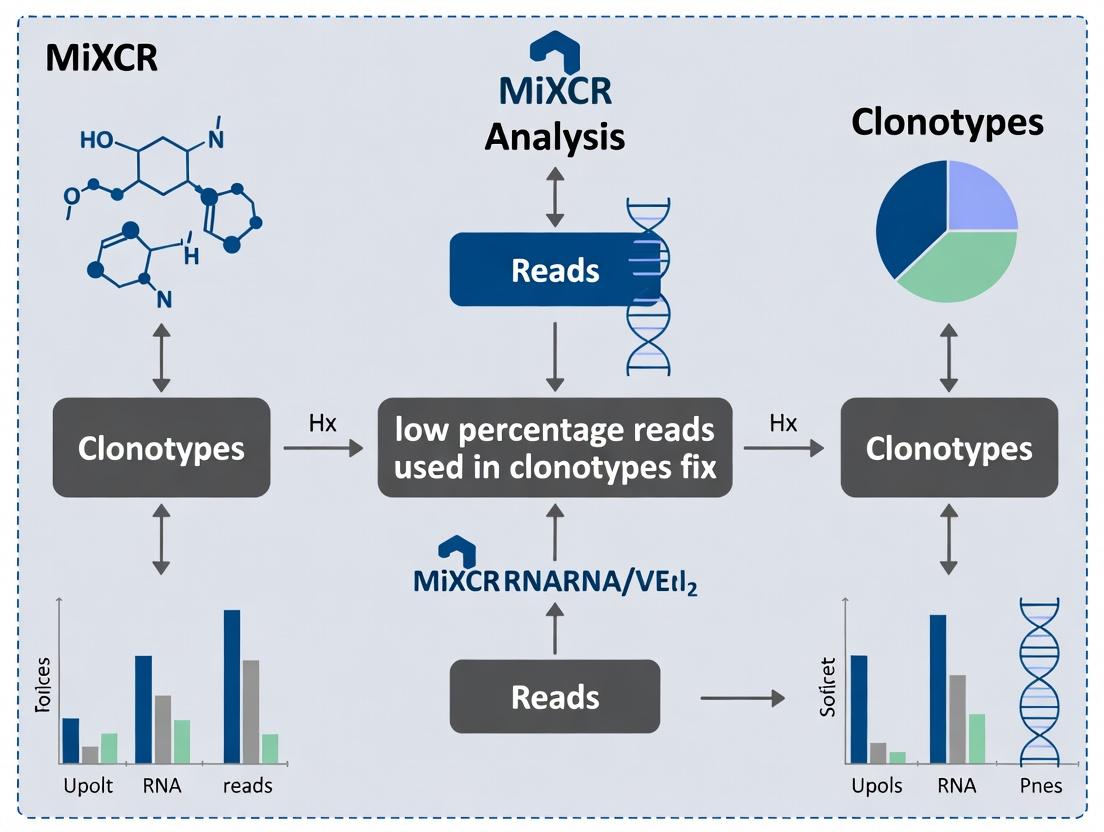
Within the context of thesis research focused on resolving low percentage reads used in clonotypes, the precise configuration of MiXCR's assembly parameters is critical. Parameters --assemble--only-mapped, --assemble--by-reads, and --assemble--collapse directly impact sensitivity, specificity, and the final clonotype repertoire structure, influencing downstream analyses in immunogenetics and drug development.
Troubleshooting Guides & FAQs
Q1: After using --assemble--only-mapped, my final clonotype count is extremely low, missing known low-frequency clones. What went wrong?
A: This parameter restricts assembly to reads that have been successfully aligned (--only-mapped), excluding all non-mapped reads from the assembly step. In samples with lower sequencing quality or high diversity, this can aggressively filter out legitimate sequences.
- Troubleshooting Protocol:
- Check the
.vdjcafile after thealignstep usingmixcr exportAlignments. Examine the percentage of aligned reads. - If alignment rate is low (<70%), revisit alignment parameters (e.g.,
--parameters rna-seqor--parameters generic) or quality-trim inputs. - Run
assemblewithout--only-mappedand compare clonotype lists. Clonotypes present only in the latter run represent the sensitive, low-frequency range sacrificed for specificity.
- Check the
Q2: When should I use --assemble--by-reads versus the default consensus assembly?
A: Use --assemble--by-reads when analyzing data from techniques like single-cell RNA-seq (e.g., 10x Genomics) or any protocol where the linkage between reads from the same original molecule must be preserved.
- Troubleshooting Protocol: If your experiment is bulk RNA/DNA-seq, do not use this flag. Using it on bulk data will create artificial, read-based clonotypes, inflating diversity and fragmenting true clones. For single-cell data, this flag is often mandatory to correctly assemble paired V and J transcripts from the same cell barcode.
Q3: My collaborator's results show a dominant clone at 15%, but my analysis with --assemble--collapse by-sequence shows it at 12%. Why the discrepancy?
A: The --collapse parameter dictates how similar sequences are grouped into a clonotype. by-sequence groups only exact nucleotide matches. by-sequence-similarity (with --min-sum-score) merges sequences based on homology, which is more biologically realistic for accounting for PCR/sequencing errors.
- Experimental Comparison Protocol:
- Run
assembletwice: once with--collapse by-sequence, once with--collapse by-sequence-similarity --min-sum-score <SCORE>(start with default of 55). - Export clonotypes (
exportClones) from both runs. - Compare the frequencies of top clones. Clones with higher frequency in the similarity-collapsed run indicate consolidation of error variants.
- Run
Table 1: Parameter Impact on Clonotype Recovery in Low Percentage Context
| Parameter | Primary Function | Advantage for Low-% Reads | Risk for Low-% Reads | Recommended Use Case |
|---|---|---|---|---|
--only-mapped |
Uses only pre-aligned reads | Reduces noise; faster assembly. | High risk of losing true low-frequency clones. | High-quality, deep bulk sequencing; initial noise-filtering runs. |
--by-reads |
Assembles from individual reads vs. consensus | Preserves low-frequency unique rearrangements from single cells. | Inflates diversity in bulk data; computationally heavier. | Mandatory for single-cell (e.g., 10x) data. Avoid for bulk. |
--collapse by-sequence |
Exact nucleotide matching | Simple, reproducible grouping. | Fragments clones into error variants, undercounting true frequency. | Rare; only for ultra-high-fidelity (e.g., PacBio HiFi) data. |
--collapse by-sequence-similarity |
Groups similar sequences (homology) | Robust consolidation of PCR/seq errors; better frequency estimation. | May over-merge highly similar but distinct clones (tune with score). | Default for most bulk NGS (Illumina); tune --min-sum-score. |
Table 2: Example Workflow Comparison for Thesis Research
| Step | Standard Sensitivity-Focused Workflow (Bulk NGS) | High-Specificity Workflow (Noisy Data) | Single-Cell Workflow (10x) |
|---|---|---|---|
| Align | mixcr align ... |
mixcr align ... --report report_align.txt |
mixcr analyze shotgun ... |
| Assemble | mixcr assemble ... |
mixcr assemble --only-mapped ... |
mixcr assemble --by-reads ... |
| Collapse | ... --collapse by-sequence-similarity |
... --collapse by-sequence-similarity |
... --collapse by-sequence-similarity |
| Goal | Maximize recovery of low-frequency clones. | Reduce false positives; focus on high-confidence clones. | Correctly pair chains per cell barcode. |
Experimental Protocols
Protocol 1: Benchmarking Parameter Impact on Low-Abundance Clones
Objective: Quantify how --only-mapped and --collapse parameters affect the recovery of spiked-in or known low-frequency clonotypes.
Materials: High-depth bulk TCR-seq data, known synthetic TCR control spike-ins (e.g., from Adaptive Biotechnologies).
Method:
- Data Preparation: Use a dataset with known low-frequency spike-in clones or subset a high-frequency clone to a known, low percentage computationally.
- Parallel Assembly: Run multiple
mixcr assemblejobs varying only the target parameter:- Job A: Default (no
--only-mapped). - Job B: With
--assemble--only-mapped. - Job C: With
--collapse by-sequence. - Job D: With
--collapse by-sequence-similarity --min-sum-score 55. - Job E: With
--collapse by-sequence-similarity --min-sum-score 70.
- Job A: Default (no
- Analysis: For each run, export clonotypes (
mixcr exportClones). Calculate the recovery rate of known low-frequency clones and the coefficient of variation (CV) of their measured frequencies across technical replicates.
Protocol 2: Resolving Single-Cell Data with--by-reads
Objective: Correctly assemble paired V(D)J sequences from 10x Genomics Chromium data.
Materials: Raw FASTQ files from a 10x V(D)J library (libraries: VDJ and GEX).
Method:
- Integrated Analysis: Use the preset
analyzecommand which automatically applies correct parameters. - Inspect Internals: The
shotgunpreset automatically invokes--assemble--by-readsduring theassemblestep. Verify this in the generated report file (sample_output.report). - Export per Cell: Export clonotypes with cell barcodes:
mixcr exportClones --chains "TRA,TRB" --prepend-attr "cellId,barcodeUMI" sample_output.clna sample_output.clones.txt.
Visualization: MiXCR Assembly Parameter Decision Pathway
Title: MiXCR Assembly Parameter Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Context of MiXCR Parameter Tuning |
|---|---|
| Synthetic TCR/Rearrangement Spike-ins | Known, low-abundance sequences used as internal controls to benchmark recovery rates of different assemble parameters. |
| High-Quality Reference Genome | Species-specific reference (e.g., GRCh38 for human) critical for accurate alignment, which directly impacts the --only-mapped filter. |
| UMI (Unique Molecular Identifier) Kits | Allows for error correction and precise quantification. When UMIs are present, the --collapse logic works on UMI consensus sequences, not raw reads. |
| Benchmarking Software (e.g., ARResT/Interrogate) | Used to compare clonotype lists generated by different parameter sets, calculating overlap and divergence metrics. |
| High-Performance Computing (HPC) Resources | Parameter testing (esp. --by-reads and similarity collapse) requires multiple parallel jobs; HPC enables rapid iteration. |
Troubleshooting Guides & FAQs
Q1: My MiXCR analysis report shows many low-abundance clones with "0" or minimal reads. Are these real clonotypes or technical artifacts?
A: Clones with very low read counts (e.g., 1-5 reads) can be challenging to interpret. They may represent genuine, rare T-cell or B-cell clones, but they can also arise from PCR errors, sequencing errors, or index hopping. It is critical to cross-reference with the cloneFraction and readCount metrics. A best practice is to set a rational threshold for filtering (e.g., a minimum of 3-5 reads per clone) and to replicate the experiment to see if the low-abundance clone appears consistently.
Q2: The "fraction" for my low-abundance clone of interest seems inconsistent between runs. How should I interpret this?
A: The cloneFraction is highly sensitive to the total read depth and the expansion of dominant clones. For low-abundance clones, the reported fraction can show significant technical variability. Focus on the absolute readCount or count (UMI-corrected count) for more stable comparison. Normalization using spike-in controls or total cell input is recommended for cross-sample comparison of low-frequency clones.
Q3: How reliable are the V/J gene alignments for clones with very few supporting reads?
A: MiXCR assigns V and J genes based on alignment scores. For clones with low reads, these assignments are less confident. Check the alignmentScore and targetSequences fields in the detailed report. A clone supported by a single read with a mediocre alignment score should be treated as a low-confidence call. Consider using the -OallowPartialAlignments=true parameter with caution, and always review the alignments in the clna or clns file with a viewer.
Q4: When using UMI-based protocols, how do I differentiate a true low-abundance clone from a UMI-collision or amplification bias?
A: UMI deduplication (-Xpreproc.umi.barcodeAttribute) is designed to mitigate PCR bias. A true low-abundance clone should have multiple reads linked to multiple unique UMIs. Review the umiCount metric. A clone with a readCount of 10 but a umiCount of 1 is highly suspect and likely an artifact. The consensus building step is crucial for accurate low-abundance quantification.
Q5: What is the best export format from MiXCR to investigate the sequence quality of low-percentage clones?
A: For deep troubleshooting, export the clone set in the cloneSeq (.clns) or cloneData (.clna) formats. These retain alignment information. You can also use the exportClones command with the -readIds or -sequence options to output the specific reads supporting a low-abundance clone for manual inspection in a tool like Geneious or IGV.
| Metric | Description | Interpretation for Low-Abundance Clones | Typical Threshold for Reliability |
|---|---|---|---|
readCount |
Total number of reads assigned to the clone. | Primary indicator of clone size. Highly variable for very low counts. | ≥ 3-5 reads (protocol-dependent). |
count (UMI) |
Number of unique molecular identifiers (UMIs) after deduplication. | Best estimate of original molecule count. More robust than readCount. |
≥ 2 unique UMIs provides higher confidence. |
cloneFraction |
Proportion of the clone relative to the total sequenced repertoire. | Very unstable metric for rare clones; sensitive to changes in dominant clones. | Use with caution. Prefer absolute counts. |
V/J Alignments |
Assigned Variable and Joining genes, with alignment scores. | Confidence decreases with low readCount. Check alignment scores. |
Score should be > 90% of the theoretical maximum. |
alignmentScore |
Numerical score from MiXCR's aligner. | Low scores indicate poor or ambiguous V/J assignment. | Compare to scores of high-confidence clones in the same sample. |
umiCount |
(UMI protocols only) Number of distinct UMIs. | Key to distinguish true rare clone from PCR duplicate artifact. | A true clone should have umiCount ≥ 1 and readCount/umiCount < 10 (approx.). |
Experimental Protocol: Validating Low-Abundance Clones
Title: Protocol for Replication and Validation of Rare Clonotypes.
Objective: To distinguish genuine low-abundance immune clones from technical noise.
Materials: See "Research Reagent Solutions" table below.
Methodology:
- Sample Splitting: Split the original RNA/cDNA sample into two or more technical replicates prior to the library preparation step.
- Independent Processing: Process each replicate through the full experimental pipeline independently (PCR, sequencing library prep).
- Independent MiXCR Analysis: Analyze each replicate with MiXCR using identical parameters (e.g.,
mixcr analyze ...). - Cross-Replicate Comparison: Identify clones present in the low-abundance range (e.g., readCount < 10) in the primary sample.
- Validation Criteria: A low-abundance clone is considered validated if it appears (with the same CDR3 amino acid sequence) in at least two independent technical replicates. The use of UMIs significantly strengthens this validation.
- Data Integration: For final analysis, merge validated clones from replicates, summing their
umiCountfor a more accurate estimate.
Visualization: Low-Abundance Clone Analysis Workflow
Diagram Title: Workflow for Analyzing Low-Abundance Clones
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Low-Abundance Clone Research |
|---|---|
| UMI-Adapters | Unique Molecular Identifiers (UMIs) are short random sequences added during library prep to each original molecule. They enable precise PCR duplicate removal and accurate quantification of rare clones. |
| High-Fidelity PCR Mix | Polymerase mixes with proofreading activity are essential to minimize PCR errors that can create artificial low-abundance sequence variants mistaken for true clones. |
| Spike-in Control Libraries | Known, synthetic immune receptor sequences added at defined, low concentrations to the sample. They act as an internal control to assess sensitivity, quantification accuracy, and detection limits for rare clones. |
| Dual-Indexing Primers | Use unique dual indices for each sample to severely reduce the risk of index hopping (crosstalk), which can misassign reads and create false, low-count clones in samples. |
| RNA/CDNA Stabilization Reagents | Preserve sample integrity from collection to processing to prevent degradation that can disproportionately affect rare transcripts and skew the perceived repertoire. |
Troubleshooting Guides & FAQs
FAQ 1: After running MiXCR, my clonotype table contains many low-frequency clones. How do I reliably export only the rare sequences (e.g., <0.01% frequency) for downstream analysis?
Answer: This is a common challenge in clonotype fix research focusing on low-abundance, potentially therapeutic clones. MiXCR's standard export includes all clonotypes. To filter for rare sequences, you must use a post-analysis filtering step. The most robust method is to use the mixcr exportClones command with the -c (chain) and -o (output) flags, followed by a custom filter based on the cloneFraction column.
Detailed Protocol:
- Export the full clonotype table:
- Filter for rare clones using a scripting tool (e.g., AWK):
This command creates a new file (
rare_clones.tsv) containing only clones with a fraction ($3) less than 0.01% (0.0001). It also exports key columns: ID, count, fraction, and CDR3 nucleotide sequence.
FAQ 2: My filtered list of rare clones is still too large. What additional filtering strategies can I apply to increase confidence in their biological relevance?
Answer: Filtering by clone fraction alone may not be sufficient. You should implement a multi-parameter export and filtering strategy to enrich for high-quality, functionally relevant rare sequences.
Detailed Protocol: Multi-Parameter Filtering Workflow
- Export a comprehensive table with additional quality metrics:
- Apply sequential filters (e.g., using R or Python) as shown in the table below:
Table 1: Sequential Filters for Rare Clonotype Analysis
| Filter Parameter | Typical Threshold | Purpose | Command/Logic Snippet (R) |
|---|---|---|---|
| Clone Fraction | < 0.01% (0.0001) | Isolate rare sequences. | df_rare <- subset(detailed_df, cloneFraction < 0.0001) |
| Consensus Quality | ≥ 30 | Ensure base calling accuracy. | df_qual <- subset(df_rare, minQualConsensus >= 30) |
| Productive Rearrangement | TRUE | Keep only in-frame, no-stop-codon sequences. | df_prod <- subset(df_qual, grepl("^[ACGT]*$", nSeqCDR3) & nchar(nSeqCDR3) %% 3 == 0) |
| V/J Gene Assignment Score | > 50% | Ensure reliable gene annotation. | df_vj <- subset(df_prod, allVHitsWithScore > 50 & allJHitsWithScore > 50) |
| CDR3 Length | 10-20 aa | Focus on canonical size range for functionality. | df_len <- subset(df_vj, nchar(aaSeqCDR3) >=10 & nchar(aaSeqCDR3) <=20) |
FAQ 3: How can I export and visualize the clonal relationship between my filtered rare clones and the dominant clones for a publication figure?
Answer: You need to export data compatible with network or phylogenetic tree visualization software. This involves exporting the nucleotide or amino acid sequences of the filtered clones alongside the top clones.
Detailed Protocol:
- Export FASTA sequences of filtered rare clones and top 50 clones:
(Create
rare_ids.txtas a tab-separated file mappingcloneIdto a descriptive name, e.g.,12345 Rare_Clone_A) - Combine FASTA files and perform multiple sequence alignment (MSA) using ClustalOmega or MAFFT.
- Generate a phylogenetic tree (e.g., via Neighbor-Joining method) and visualize it in FigTree or iTOL.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Rare-Sequence Clonotype Analysis
| Item | Function in Analysis |
|---|---|
| MiXCR Software Suite | Core tool for aligning reads, assembling clonotypes, and initial quantification from NGS immune repertoire data. |
| High-Fidelity PCR Master Mix | Critical for library prep to minimize PCR errors that create artificial rare sequences. |
| UMI (Unique Molecular Identifier) Adapters | Enables correction for PCR and sequencing errors, distinguishing true rare clones from technical artifacts. |
| SPRIselect Beads | For precise size selection and clean-up during NGS library preparation to maintain diversity. |
| R/Bioconductor (edgeR, immunarch) | Statistical environment for advanced differential abundance testing and repertoire analysis post-export. |
| Python (Pandas, SciPy) | For building custom filtering, analysis pipelines, and data visualization scripts. |
| Phylogenetic Tree Visualization Tool (e.g., FigTree) | To illustrate evolutionary relationships between rare and dominant clones. |
Visualizations
Diagram Title: Rare Clone Filtering and Analysis Workflow
Diagram Title: Command-Line Pipeline for Rare Sequence Export
Diagnosing Pitfalls: Solutions for PCR Errors, False Positives, and Data Loss in MiXCR Analysis
FAQs & Troubleshooting Guides
Q1: When analyzing MiXCR output, I see many low-abundance clonotypes (<0.1%). How can I determine if these are genuine rare clones or PCR/sequencing errors? A: This is a core challenge. Low-frequency reads can originate from true biological diversity (e.g., a nascent immune response) or from artifacts introduced during library prep and sequencing. Follow this decision tree and protocol.
Experimental Protocol 1: Distinguishing Artifacts from True Diversity via Technical Replicates
- Sample Splitting: Split your cDNA sample into at least 3 independent PCR amplification reactions before adding sample-specific barcodes.
- Independent Library Prep: Process each replicate through separate library preparation steps (including indexing).
- Sequencing: Pool and sequence replicates on the same flow cell to minimize run-to-run variability.
- MiXCR Analysis: Analyze each replicate independently with MiXCR using identical parameters (
align,assemble). - Cross-Replicate Comparison: True biological clonotypes are more likely to appear in multiple technical replicates. Artifacts (e.g., polymerase errors, tag jumps) are typically stochastic and replicate-specific.
Table 1: Interpretation of Low-Frequency Clonotypes Across Technical Replicates
| Clonotype Detection Pattern | Likely Origin | Recommended Action |
|---|---|---|
| Present in all 3+ replicates | High-confidence true clone | Include in downstream analysis. |
| Present in 2 out of 3 replicates | Probable true clone | Consider including; review alignments. |
| Present in only 1 replicate | Probable PCR/sequencing artifact | Filter out using MiXCR's -c (minimal clone count) or downstream analysis. |
Q2: What are the main PCR artifacts that inflate clonotype diversity, and how can I mitigate them? A: Key artifacts include:
- Polymerase Errors: Nucleotide misincorporation during amplification.
- Chimeras/Template Switching: Hybrid amplicons from co-amplified fragments.
- PCR Bottlenecking (Uneven Amplification): Stochastic primer binding causing false skew in abundance.
Experimental Protocol 2: Mitigating PCR Artifacts with Enzyme & Protocol Selection
- Use High-Fidelity Polymerase: Utilize polymerases with proofreading activity (e.g., Q5, KAPA HiFi). This reduces point mutation artifacts.
- Limit PCR Cycles: Use the minimum number of PCR cycles necessary for library construction to reduce chimera formation and error accumulation.
- Employ Unique Molecular Identifiers (UMIs): Incorporate UMIs during reverse transcription. This is the gold standard.
- Protocol: During cDNA synthesis, use primers containing random UMI sequences. After PCR and MiXCR analysis, the software can collapse reads originating from the same original mRNA molecule (
exportClones --umi-count-collection), correcting for PCR duplicates and many polymerase errors.
- Protocol: During cDNA synthesis, use primers containing random UMI sequences. After PCR and MiXCR analysis, the software can collapse reads originating from the same original mRNA molecule (
Q3: My negative control (no template) shows clonotypes after MiXCR analysis. What does this indicate? A: This signals significant contamination or index hopping (also known as tag jumping).
Troubleshooting Steps:
- Review Wet-Lab Practices: Ensure strict separation of pre- and post-PCR areas. Use dedicated equipment and aerosol-resistant filter tips.
- Check for Index Hopping: This is prevalent on patterned flow cell sequencers (e.g., Illumina NovaSeq).
- Solution: Use dual indexing (unique i5 and i7 indices per sample). MiXCR can filter some cross-talk, but the primary solution is using unique dual indexes during library prep to bioinformatically identify and remove mis-assigned reads.
- Filter in MiXCR: Use the
-c(minimal clone count per sample) parameter aggressively for negative controls. Any clonotype passing this filter in a negative control is a high-priority contaminant candidate.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for High-Fidelity Immune Repertoire Profiling
| Item | Function | Key Consideration |
|---|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, KAPA HiFi) | Amplifies template with minimal nucleotide misincorporation. | Critical for reducing point mutation artifacts that mimic somatic hypermutation. |
| UMI-linked RT Primers | Contains random molecular barcodes to tag each original mRNA molecule. | Enables correction for PCR duplication and estimation of pre-amplification molecule count. |
| Unique Dual Index Kit (e.g., Illumina) | Provides unique i5 and i7 index combinations for each sample. | Mitigates index hopping artifacts on high-throughput sequencers. |
| RNase Inhibitor | Protects RNA template from degradation during reverse transcription. | Preserves the original RNA population, preventing loss of low-abundance transcripts. |
| Magnetic Beads (SPRI) | For size selection and clean-up. | Removes primer dimers and large contaminants; critical for maintaining library quality. |
Workflow for Validating Low-Abundance Clonotypes
Pathway of PCR Artifact Generation
Troubleshooting Guides & FAQs
Q1: During MiXCR analysis, my final clonotype table is missing clones that appeared in intermediate alignment files. Why do these low-abundance clones "disappear"? A: This is typically a result of MiXCR's built-in quality and error-correction filters. The primary mechanisms are:
- Quality Filtering: Reads with low base quality scores are discarded during alignment to prevent false clonotype calls from sequencing errors.
- Error Correction & Clustering: MiXCR groups similar sequences, assuming minor differences are PCR or sequencing errors. Low-count sequences are often merged into a dominant, high-count neighbor if they fall within the specified clustering threshold.
- Abundance Thresholds: The most direct cause. The
--minimal-clone-countand--minimal-frequencyparameters set absolute and relative abundance cutoffs. Clones failing to meet these are excluded from the final report to focus on robust, reproducible signals and reduce noise.
Q2: How can I recover these clones for my clonotypes fix research without compromising data integrity? A: A balanced approach is required:
- Adjust Parameters: Relax the
--minimal-clone-count(e.g., from default 2 to 1) and--minimal-frequencyparameters cautiously. This will increase sensitivity but also noise. - Modify Clustering: Increase the
--cluster-for-error-correctionthreshold (e.g., from default 0.01 to 0.005) to make merging less aggressive. Consider using the--no-error-correctionflag for exploratory analysis, but be aware this will report all raw sequencing variants. - Post-Processing: Export the full, unfiltered clonotype list using
--verboseor--no-filter-resultsand apply custom filters later using R or Python, allowing you to track all clones through your analysis pipeline.
Q3: What are the trade-offs between sensitivity (keeping low-percentage clones) and specificity (avoiding false positives)? A: The core trade-off is data reliability versus comprehensiveness.
| Parameter/Strategy | Goal: Increase Sensitivity (Keep More Clones) | Goal: Increase Specificity (Reduce Noise) | Primary Risk |
|---|---|---|---|
| Minimal Clone Count | Decrease value (e.g., to 1) | Increase value (e.g., to 3 or 5) | More false positives from sequencing errors / Fewer true low-abundance clones |
| Error Correction | Less aggressive (higher threshold) or disabled | Default or more aggressive (lower threshold) | Inflated diversity from errors / Merging of biologically distinct low-count clones |
| Quality Filtering | Relax PHRED score requirements | Strict PHRED score requirements | Higher error rate in aligned reads / Loss of data from lower-quality runs |
Detailed Methodology: Investigating Low-Abundance Clonotypes
Protocol: Controlled Spike-in Experiment for Sensitivity Threshold Validation
- Spike-in Design: Synthesize known, low-abundance TCR or BCR sequences at defined ratios (e.g., 0.01%, 0.05%, 0.1%) within a background of polyclonal cDNA.
- Library Preparation & Sequencing: Process the spike-in sample alongside a pure polyclonal control using your standard NGS workflow (e.g., 5' RACE, UMIs). Use sufficient sequencing depth (>100,000 reads per sample).
- Dual-Pipeline Analysis:
- Pipeline A (Standard): Analyze with laboratory-standard MiXCR parameters (e.g.,
--minimal-clone-count 2). - Pipeline B (Sensitive): Analyze with relaxed parameters (
--minimal-clone-count 1 --minimal-frequency 0).
- Pipeline A (Standard): Analyze with laboratory-standard MiXCR parameters (e.g.,
- Recovery Assessment: In the final clonotype tables, calculate the recovery rate of each spike-in clone as
(Observed Count / Expected Count) * 100. This quantifies the detection limit of your current workflow.
Protocol: UMI-Based Validation of Low-Percentage Clones
- Experimental Setup: Use a UMI (Unique Molecular Identifier)-based library prep kit. This allows distinction between true biological molecules and PCR duplicates/errors.
- MiXCR Analysis: Run MiXCR with the
--umiflag enabled for correct UMI-based clustering and error correction. - Validation Criterion: A low-abundance sequence supported by multiple independent UMIs is highly likely to be a true biological clone, not an artifact. This provides a robust basis for retaining it in the final analysis, even if its read percentage is low.
Visualizations
Title: MiXCR Filtering Pipeline Where Low-Percentage Reads Are Lost
Title: UMI-Based Validation Workflow for Low-Abundance Clones
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Clonotype Fix Research |
|---|---|
| UMI-Compatible cDNA Synthesis Kit | Embeds unique molecular identifiers during reverse transcription to tag original mRNA molecules, enabling distinction between PCR duplicates and true biological diversity. Critical for validating low-count clones. |
| Multiplex PCR Primers (TRB/IGHV) | Amplifies the variable region of T-cell or B-cell receptors. High-quality, validated primers ensure balanced representation of the repertoire, minimizing amplification bias that can obscure low-percentage clones. |
| Spike-in Synthetic Clonotype Controls | Artificially synthesized TCR/BCR sequences at known, low concentrations. Used as internal controls to empirically determine the detection limit and recovery efficiency of the wet-lab and bioinformatics pipeline. |
| High-Fidelity DNA Polymerase | Enzyme with proofreading capability for PCR amplification. Essential for minimizing introduced errors during library prep, which can create artificial variants mistaken for low-abundance true clones. |
| MiXCR Software Suite | The core analysis pipeline for bulk immune repertoire sequencing. Its parameters (--minimal-clone-count, clustering thresholds) are the primary tools for managing the sensitivity/specificity balance. |
The Role of UMIs (Unique Molecular Identifiers) in Validating Rare Clonotypes
Troubleshooting Guides & FAQs
Q1: After UMI-based error correction in MiXCR, my final repertoire shows zero or extremely low counts for previously identified low-percentage clonotypes. What went wrong? A: This is a critical validation step. The likely cause is that those initial "clonotypes" were PCR/sequencing artifacts. UMIs collapse PCR duplicates and correct sequencing errors, revealing true biological molecules. To troubleshoot:
- Check your UMI length and complexity. Short UMIs (<8bp) may not provide sufficient diversity to tag all molecules, leading to collisions and over-correction.
- Analyze the
clones.txtfile columnuniqueUmisCountorumiCount. A true rare clonotype will have at least 2-3 reads supported by different UMIs. - Verify UMI extraction and alignment during
mixcr analyzewith the--verboseflag. Ensure UMIs are not corrupted by low-quality bases at the start of reads.
Q2: How do I determine the optimal threshold for filtering by UMI count when validating rare clonotypes? A: There is no universal threshold; it is experiment-dependent. You must perform a titration analysis.
| UMI Count Threshold | Clonotypes Reported | Likely False Positives | Risk Profile |
|---|---|---|---|
| ≥ 1 | Maximum | Very High | Includes all PCR/sequencing errors. Not validated. |
| ≥ 2 | High | Moderate | Common starting point; validates molecules with ≥2 independent copies. |
| ≥ 3 | Conservative | Low | High confidence for rare variants in bulk sequencing. |
| Statistical (Poisson) | Data-Driven | Controlled | Uses read/UMI models to estimate noise floor (see MiXCR -c options). |
Protocol for Threshold Titration:
- Run MiXCR with UMI correction:
mixcr analyze ... --starting-material rna --receptor-type trb --umi .... - Export the clonotype table.
- Filter the
clones.txtfile iteratively, requiringuniqueUmisCount≥ 1, ≥ 2, ≥ 3. - Plot the number of clonotypes retained vs. the threshold. The curve often shows an inflection point where noise is removed. Validate biologically with a known positive control.
Q3: I am getting "No UMIs found" warnings during processing. How do I fix my FASTQ files? A: This indicates a mismatch between your data's UMI location and the command parameters.
- Issue: UMIs are in a separate read (R3) but you used
--umi-position R1. - Fix: Use
--umi-position R3. - Issue: UMIs are embedded in the R1 read (e.g., first 10bp) but you didn't specify the length.
- Fix: Use
--umi-position prefix --umi-length 10. - General Protocol: Always inspect your raw FASTQ files with a tool like
seqtkto confirm UMI location:seqtk seq -A your_file_R1.fastq | head -2.
Q4: Can UMI analysis in MiXCR help correct for low-template PCR bias in single-cell or low-input samples? A: Yes, this is a primary strength. UMIs allow absolute molecule counting, mitigating amplification bias. However, for very low inputs, follow this:
- Increase UMI Complexity: Use longer UMIs (10-12bp).
- Adjust Pipeline: Use
mixcr analyzewith the--downsampling noneoption to prevent stochastic loss of rare clones. - Filter Cautiously: Start with a UMI count threshold of ≥2. Calculate the "molecules per cell" or "molecules per input mass" using the UMI count, not the read count.
The Scientist's Toolkit: Key Reagent Solutions
| Item | Function in UMI/Clonotype Validation |
|---|---|
| UMI-equipped Adapters (e.g., NEBNext) | Integrates random molecular barcodes during cDNA library prep, enabling molecule-level tracking. |
| High-Fidelity DNA Polymerase (e.g., Q5, KAPA HiFi) | Minimizes PCR introduction of errors that could create artificial clonal diversity, critical before UMI deduplication. |
| SPRIselect Beads (Beckman Coulter) | For precise size selection and clean-up post-library prep, removing adapter dimers that consume sequencing depth. |
| MiXCR Software Suite | Integrated pipeline for align, assemble, and export with built-in UMI handling, error correction, and clone quantification. |
| Unique Dual Indexes (UDIs) | Used in conjunction with UMIs to further eliminate index hopping (crosstalk) artifacts between samples on high-throughput sequencers. |
Experimental Workflow & Data Relationships
Diagram Title: UMI-Based Validation Workflow for Rare Clonotypes
Diagram Title: How UMIs Collapse PCR Duplicates and Correct Errors
Benchmarking Accuracy: How MiXCR's Rare Clone Detection Compares to Alternative Tools
Troubleshooting Guides and FAQs
Q1: I am using MiXCR for my TCR-seq data, but the final clonotype report shows a very low percentage of reads assigned (< 20%). What are the primary causes and how can I fix this?
A: Low assigned read percentages in MiXCR are common in low-quality or highly complex samples. Key causes and solutions:
- Cause 1: Low RNA Input/Sequencing Depth. The library may be dominated by non-immune or non-productive transcripts.
- Fix: Ensure sufficient input RNA (≥100 ng total RNA) and target ≥50,000 reads per sample for repertoire studies. Use target enrichment (e.g., multiplex PCR for V genes).
- Cause 2: Poor Read Quality or Adapter Contamination.
- Fix: Run
mixcr analyze shotgunwith the--only-productiveflag after theassemblestep. Pre-trim adapters usingmixcr analyze shotgun --starting-material rna --5-end no-v-primers --3-end c-primers. Check raw FASTQC reports.
- Fix: Run
- Cause 3: Incomplete Reference Library for your organism or chain type.
- Fix: Use the
--speciesflag explicitly (e.g.,--species hsa). Check and, if needed, customize the V/D/J/C gene reference database using themixcr importSegmentscommand.
- Fix: Use the
Q2: How do I increase the sensitivity of MiXCR for detecting rare clones without generating excessive false positives?
A: Sensitivity is tuned primarily in the align and assemble steps.
- Protocol Adjustment: Use
mixcr align --report {file} --save-reportto inspect alignment metrics. Increase the-OallowPartialAlignments=trueparameter for challenging data. In theassemblestep, carefully adjust:--minimal-score(default 150). Lowering to 120 increases sensitivity but may add noise.--minimal-sum-qualities(default 0). Set to 30-50 to filter low-confidence alignments.- The recommended workflow for sensitive assembly of rare clones is:
mixcr analyze shotgun --assemble "-OseparateByC=true -OseparateByV=true -OseparateByJ=true --minimal-score 130 --minimal-sum-qualities 40" sample_R1.fastq.gz sample_R2.fastq.gz result
Q3: When comparing MiXCR to IgBLAST/IMGT, I get different dominant clonotypes. Which tool is correct?
A: Discrepancies are expected due to fundamental algorithmic differences.
- Root Cause: MiXCR uses a unified, overlapping k-mer-based aligner and a clustering-based assembler. IgBLAST uses BLAST against a germline database. IMGT/HighV-QUEST is highly standardized but less flexible.
- Troubleshooting Protocol:
- Validate Alignment: Extract the raw reads for the disputed clonotype from each tool's output. Manually inspect the alignment in a tool like IGV.
- Check Parameters: Ensure equivalence in key parameters: clustering threshold (MiXCR's
--minimal-score), germline reference version, and the definition of a "productive" sequence. - Benchmark with Spiked-in Controls: If possible, validate using a synthetic repertoire with known clones.
Q4: What are the main advantages of using TRUST4 for tumor-infiltrating lymphocyte (TIL) data, and when should I prefer it over MiXCR?
A: TRUST4 is specifically designed for unsorted RNA-Seq or Whole Exome/Genome Sequencing data.
- Use TRUST4 when: Your input is bulk tumor RNA-seq (no TCR enrichment), as it can extract and assemble TCR/IG sequences directly from background transcriptomic data. It is robust to high levels of somatic mutation.
- Use MiXCR when: You have targeted immune repertoire sequencing data (e.g., from 5'RACE or multiplex PCR). MiXCR generally provides more precise quantitation and finer clustering for enriched libraries.
- Integration Fix: For TIL studies within a broader thesis, a robust protocol is to run both pipelines. Use TRUST4 for de novo identification of clones from bulk data, then use the clonotypes as a reference to guide a more sensitive re-analysis of enriched data with MiXCR.
Table 1: Core Algorithmic & Practical Comparison
| Feature | MiXCR | IgBLAST | IMGT/HighV-QUEST | TRUST4 |
|---|---|---|---|---|
| Core Algorithm | Overlapping k-mer alignment, clustering assembler | BLAST-based local alignment | Standardized, rule-based alignment | De novo assembly from RNA-seq, BLAST-like alignment |
| Input Data | Fastq (paired/single), BAM | Fastq, FASTA | FASTA only (length-restricted) | Fastq, BAM (Standard RNA-seq) |
| Speed | Very Fast | Moderate | Slow (queue-based) | Moderate-Slow |
| Customization | High (adjustable at each step) | Moderate | Low | Low-Moderate |
| Best Use Case | High-throughput targeted repertoire data | Standardized single-sequence analysis, compatibility | Publication-standard annotation, germline comparison | Extracting repertoires from bulk RNA/WXS/WGS |
| Key Limitation | Can be sensitive to initial parameters | Less accurate for hypermutated sequences | Rigid input format, slow turnaround | Less precise quantitation for enriched libraries |
Table 2: Typical Performance on Low-Input/Complex Samples (Thesis Context)
| Metric | MiXCR | IgBLAST | IMGT/HighV-QUEST | TRUST4 |
|---|---|---|---|---|
| % Reads Assigned | 60-95%* (on target-enriched data) | 50-80% | 50-75% | 1-10% (of total RNA-seq reads) |
| Rare Clone Sensitivity | High (tunable) | Moderate | Moderate | Very High (in context of bulk data) |
| Handling of Somatic Hypermutation | Good | Poor | Excellent | Excellent |
| Ease of Reproducibility | High (single command pipelines) | Moderate | High (fixed parameters) | Moderate |
*Low percentage (<20%) indicates need for troubleshooting (see FAQ 1).
Experimental Protocols
Protocol 1: Benchmarking Clonotype Calling Accuracy
- Spike-in Control: Use a synthetic repertoire (e.g., Lymphocyte RNA control) with known clonotype sequences and frequencies.
- Data Processing: Process the same FASTQ files independently through MiXCR (
mixcr analyze shotgun), IgBLAST (viaMakeDb.pyin Change-O), and TRUST4 (run-trust4). - Alignment: Map the raw reads of each called clonotype back to the known reference sequences using a strict aligner (e.g., Bowtie2).
- Validation Metric: Calculate precision and recall for each tool: Precision = TP / (TP + FP), Recall = TP / (TP + FN).
Protocol 2: Integrated Analysis for Tumor Repertoire (Thesis Workflow)
- Bulk RNA-seq Analysis (TRUST4): Run
run-trust4 -f <reference> -b <bam> -o <prefix>on your tumor RNA-seq BAM file to generate an initial, unbiased clonotype list. - Targeted Repertoire Sequencing (MiXCR): Process your dedicated TCR-seq library with
mixcr analyze amplicon --starting-material dna --5-end v-primers --3-end j-primers. - Data Integration: Cross-reference the dominant clonotype sequences from the TRUST4 output with the MiXCR results. Use the MiXCR quantified frequencies for these overlapping clones as the most accurate measure.
- Validation: Extract consensus sequences and design primers for validation via Sanger sequencing or digital PCR.
Visualizations
Title: Integrated Tumor Repertoire Analysis Workflow
Title: Troubleshooting MiXCR Low Assigned Reads
The Scientist's Toolkit
| Research Reagent / Tool | Function in Repertoire Analysis |
|---|---|
| SMARTer Human TCR a/b Profiling Kit | Provides target enrichment via 5'RACE for unbiased, full-length V(D)J capture from low-input RNA. |
| Lymphocyte RNA Control (e.g., from cell lines) | Synthetic repertoire spike-in for benchmarking pipeline accuracy and sensitivity. |
| IGG or TCRG Primer Sets (Multiplex PCR) | For DNA-based library preparation from genomic DNA, covering most V genes. |
| DNeasy Blood & Tissue Kit | High-quality genomic DNA extraction for DNA-based TCR/IG library prep. |
| RNeasy Micro Kit | Reliable total RNA isolation from low-cell-number samples (e.g., fine needle aspirates). |
| MiXCR Software Suite | Integrated pipeline for end-to-end analysis, from raw reads to clonotype tables. |
| Change-O / ImmuneDB | Suite of tools for post-processing IgBLAST output and advanced repertoire statistics. |
| IMGT Reference Directories | Curated, high-quality germline V, D, J, and C gene sequences for accurate alignment. |
Troubleshooting Guides & FAQs
Q1: During MiXCR analysis of low-abundance clones, my clonotype table shows unexpected high diversity in negative controls. What could be the cause and how can I address it? A: This is often due to index hopping or cross-contamination between samples in multiplexed sequencing runs. To troubleshoot:
- Verify Library Preparation: Ensure unique dual indices (UDIs) are used to mitigate index hopping.
- Analyze Negative Controls: Process your negative control (no-template) samples through the same MiXCR pipeline. Clonotypes appearing in both test and control samples are likely artifacts.
- Apply a Threshold: Filter out clonotypes from your test samples that appear in the negative control with a count above 2-5 reads.
- Spike-In Solution: Use a synthetic immune repertoire library (e.g., from ReachBio or ImmuneCODE) as an internal negative control. Any clonotype in your sample that matches the synthetic library is a contamination artifact.
Q2: How do I determine the limit of detection for low-frequency clones (e.g., <0.01%) in my repertoire using MiXCR? A: Determining the limit of detection (LOD) requires a ground-truth standard.
- Use Spike-Ins: Employ a commercially available synthetic T-cell/B-cell receptor library with known sequences at defined, low frequencies (e.g., 0.001%, 0.01%, 0.1%).
- Experimental Protocol:
a. Spike the synthetic library into your sample DNA at the desired dilutions.
b. Perform library preparation and sequencing alongside an unspiked sample.
c. Process all files through MiXCR (
mixcr analyze amplicon). d. Align the detected clonotypes against the known spike-in sequences. - Calculate Sensitivity: For each spike-in frequency, calculate: (Number of spike-in clonotypes detected by MiXCR) / (Total number of unique spike-in clonotypes added). This directly measures MiXCR's sensitivity at that abundance level.
Q3: When using spike-ins for validation, how do I distinguish between the spike-in sequences and my biological sample during MiXCR analysis? A: You must maintain a reference list of all spike-in sequences.
- Tag Sequences: Some synthetic libraries come with unique "barcode" segments within the CDR3. You can create a custom MiXCR alignment reference that includes these barcodes.
- Post-Analysis Filtering: The most common method is to perform a BLAST or exact string match of all output CDR3 nucleotide or amino acid sequences from MiXCR against your reference spike-in list. Remove any matches.
- Dedicated Alignment: Run MiXCR separately on the sample with spike-ins using a reference that includes the spike-in V and J genes. This will explicitly annotate them.
Q4: My calculated specificity seems low. What experimental or bioinformatic steps can improve it? A: Low specificity indicates a high false discovery rate (FDR). Key actions:
- Optimize MiXCR Parameters: Adjust the
--alignand--assembleparameters, particularly-OmaxBadPointsPercentand--min-sum-freq, to be more stringent. This reduces false alignments. - Implement a UMI-based Protocol: Use Unique Molecular Identifiers (UMIs) during cDNA synthesis. MiXCR's
--use-umisoption in theassemblefunction can collapse PCR duplicates, distinguishing true low-abundance sequences from PCR/sequencing errors. - Apply Abundance Thresholds: Establish a minimum read count per clonotype (e.g., ≥3 reads) based on your negative control analysis. Clonotypes below this threshold are likely noise.
- Validate with an Alternative Tool: Run a subset of data through an independent pipeline (e.g., ImmunoSEQ, VDJPuzzle) to compare clonotype calls.
Q5: What is the best way to format and use quantitative data from spike-in experiments to validate MiXCR's performance? A: Structure your validation data into clear summary tables.
Table 1: Sensitivity Assessment Using Defined Spike-In Frequencies
| Spike-in Clonotype Frequency (%) | Number of Unique Spike-ins Added | Number Detected by MiXCR | Calculated Sensitivity (%) |
|---|---|---|---|
| 1.000 | 50 | 50 | 100.0 |
| 0.100 | 50 | 49 | 98.0 |
| 0.010 | 50 | 45 | 90.0 |
| 0.001 | 50 | 12 | 24.0 |
Table 2: Specificity Assessment Using Synthetic Library & Negative Controls
| Sample Type | Total Clonotypes Called by MiXCR | Clonotypes Matching Synthetic Library (False Positives) | Clonotypes in Negative Control | Estimated Specificity (%)* |
|---|---|---|---|---|
| Biological Sample A | 15,240 | 28 | 15 | 99.72 |
| Negative Control | 210 | 0 | 210 | N/A |
| *Specificity Calculation: (Total Clonotypes - False Positives) / Total Clonotypes x 100%. Clonotypes from the negative control are a subset of false positives. |
Detailed Experimental Protocols
Protocol 1: Sensitivity Limit of Detection (LOD) using Serial Dilution of Synthetic Clonotypes
Objective: To empirically determine the lowest clonotype frequency MiXCR can reliably detect. Materials: Biological sample gDNA, synthetic TCR/BCR library (e.g., ImmuneCODE Mirage), qPCR kit, NGS library prep kit. Steps:
- Quantify & Dilute: Precisely quantify your synthetic library and biological sample gDNA using a Qubit fluorometer.
- Create Spike-in Series: Spike the synthetic library into aliquots of biological gDNA to create a dilution series (e.g., 1%, 0.1%, 0.01%, 0.001% by molecule count). Include a no-spike control.
- Amplify & Sequence: Perform multiplex PCR for TCR/BCR loci, add sequencing adapters/indices, and pool libraries equimolarly. Sequence on an Illumina platform with sufficient depth (≥100,000 reads per sample).
- MiXCR Analysis:
- Validation: Cross-reference the
output_report.clonotypes.ALL.txtfile with the known list of spike-in sequences. Calculate sensitivity as shown in Table 1.
Protocol 2: Specificity & Background Error Rate using UMIs and Negative Controls
Objective: To quantify and minimize false positive clonotype calls. Materials: UMI-equipped adapters, synthetic immune repertoire (for false positive control), no-template control. Steps:
- Library Prep with UMIs: Perform cDNA synthesis using primers containing UMIs. Proceed with gene-specific amplification and NGS library preparation. Include a well containing only the synthetic library and a no-template water control.
- Sequencing: Use a paired-end run with sufficient length to cover the UMI.
- MiXCR Analysis with UMI Deduplication:
- Error Rate Calculation: Identify all clonotypes in the no-template and synthetic-library-only controls. Any clonotype called here is a technical artifact (false positive). The frequency of these artifacts in your test samples defines your background error rate, informing your
--min-sum-freqthreshold.
Workflow & Relationship Diagrams
Title: Ground-Truth Validation Workflow for MiXCR
Title: Troubleshooting Low % Clonotype Analysis Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Validation | Example/Supplier |
|---|---|---|
| Synthetic Immune Repertoire Library | Provides known, clonally unrelated TCR/BCR sequences as a spike-in control for sensitivity & a negative control for specificity. | Mirage Synthetic Immune Repertoire (ImmuneCODE), ReachBio Reference Standards. |
| Unique Molecular Identifiers (UMIs) | Short random nucleotides added during cDNA synthesis to tag each original molecule, enabling correction for PCR and sequencing errors. | NEBNext Unique Duplex UMI Adapters, SMARTer UMI technology. |
| Unique Dual Index (UDI) Kits | Prevents index hopping between multiplexed samples, reducing cross-sample contamination false positives. | Illumina IDT UDIs, Nextera UD Indexes. |
| Clonal Template Standard | A single known clonotype at high concentration for absolute quantification and linearity assessment. | ATCC T-cell Receptor Gene Standards. |
| No-Template Control (NTC) Reagents | The same water, enzymes, and master mix used for test samples to identify kit or environmental contamination. | Nuclease-free Water (Ambion), PCR Master Mix. |
| High-Sensitivity DNA/RNA Assay | Accurate quantification of low-input and dilute samples is critical for precise spike-in dilution series. | Qubit dsDNA HS Assay (Invitrogen), Bioanalyzer HS DNA Kit (Agilent). |
FAQs & Troubleshooting
Q1: After analyzing MiXCR-derived, low-abundance clonotype data in VDJtools, my diversity estimates (e.g., Chao1, Shannon) appear artificially high. What could be the cause and solution?
A: This often stems from insufficient filtering of technical noise from PCR/sequencing errors in the low-count reads. VDJtools' FilterNonFunctional and Downsample commands are critical first steps. Use the CalcDiversityStats function only after applying an abundance threshold (e.g., --min-reads 3). Validate by comparing the curve shape in rarefaction plots generated by CalcRarefactionCurve.
Q2: When using ImmuneSIM to generate synthetic repertoires for validation, what parameters should I match to my real MiXCR dataset to ensure a fair comparison?
A: Match these key parameters in your ImmuneSIM simulation:
- Species and Gene Loci: Set
species = "Human"andchain = "TRB"or"IGH"accordingly. - Number of Sequences: Set
number_of_seqsto match the post-filtering count of your experimental data. - Repertoire Type: Use
repertoire_type = "shm.age"for B cells or"naive"for naive T cells to approximate diversity. - Initial Clonal Expansion: Adjust
initial_clonal_expansion = "yes"andclonal_size_dist = "uniform"to model a similar clonal structure.
Q3: I receive a "Sample tags not unique" error in VDJtools when trying to merge multiple metadata files. How do I resolve this?
A: This error indicates duplicate sample identifiers in your metadata file. Ensure the sample column is unique for each row. Verify that your MiXCR output files were processed with distinct sample tags. The metadata file should be a tab-delimited text file with columns: sample, group, subject, and other optional fields.
Q4: How can I visually compare the clonal overlap between my MiXCR-derived low-percentage clonotypes and the ImmuneSIM-simulated baseline?
A: Use the VDJtools OverlapPair function on the filtered, normalized data. Then, generate a Venn diagram or a Circos plot for visualization. For a quantitative comparison, create a summary table from the overlap output.
Table 1: Quantitative Overlap Metrics Between Experimental (MiXCR) and Simulated (ImmuneSIM) Repertoires
| Metric | MiXCR Dataset A | ImmuneSIM Simulated Set | Interpretation |
|---|---|---|---|
| Total Unique Clonotypes | 15,342 | 14,850 | Comparable repertoire depth |
| Shared Clonotypes (Exact) | 127 | 127 | Core public sequences |
| Jaccard Similarity Index | 0.0082 | 0.0085 | Low overlap, as expected |
| Normalized Morisita-Horn | 0.19 | 0.19 | Low ecological similarity |
Q5: What is the recommended workflow to validate that my low-percentage MiXCR clonotypes are biologically relevant and not artifacts?
A: Follow this integrative validation protocol:
- Preprocessing: Filter MiXCR results for functional sequences and aggregate by sample using VDJtools.
- Simulation: Generate a null-model repertoire with ImmuneSIM using matched parameters.
- Comparative Analysis: Use VDJtools to compute diversity, overlap, and gene usage.
- Statistical Testing: Apply the
CalcSpectratypeandCalcSegmentUsagefunctions to both datasets and compare distributions using the provided tests (e.g., Chi-squared). - Visualization: Generate comparative plots (e.g., PCA via
CalcSegmentUsage).
Experimental Protocol: Validating Low-Abundance Clonotypes
Title: Protocol for Integrative Validation of Low-Frequency TCR/BCR Clonotypes Using MiXCR, VDJtools, and ImmuneSIM.
Purpose: To establish a pipeline for verifying the biological significance of low-percentage clonotype calls from MiXCR output.
Materials:
- MiXCR-aligned clone set files (
.clnsor text export). - VDJtools software (v1.2.1 or higher).
- ImmuneSIM R package (v1.0.0 or higher).
- R or Python environment for statistical analysis.
Procedure:
- Data Conversion: Convert MiXCR files to VDJtools format using
vdjtools convert -S mixcr. - Basic Filtering: Run
vdjtools FilterNonFunctionalto remove non-functional sequences andvdjtools Downsampleto normalize library sizes. - Noise Filtering (Critical for Low Reads): Apply a count-based filter (e.g.,
--min-reads 2) to mitigate sequencing error noise. Usevdjtools Decontaminateif batch effects are suspected. - Synthetic Repertoire Generation: In R, run ImmuneSIM:
sim_repertoire <- immuneSIM(number_of_seqs = 15000, species = "Human", chain = "TRB", repertoire_type = "naive"). Export the result for VDJtools. - Comparative Analysis:
- Diversity:
vdjtools CalcDiversityStats -m metadata.txt input.txt output/ - Gene Usage:
vdjtools CalcSegmentUsage -p -m metadata.txt input.txt output/ - Overlap:
vdjtools OverlapPair -p real_sample.txt simulated_sample.txt output/
- Diversity:
- Visualization & Interpretation: Generate and compare rarefaction curves, spectratypes, and PCA plots from the outputs.
Research Reagent Solutions
Table 2: Essential Toolkit for Immunosequencing Validation Analysis
| Item | Function | Example/Note |
|---|---|---|
| VDJtools Software | Primary toolset for post-processing, normalization, and comparative analysis of immune repertoire sequencing data. | Command-line tool. Essential for converting MiXCR outputs. |
| ImmuneSIM R Package | Generates synthetic, biologically realistic immunoglobulin/T-cell receptor repertoire sequences for negative controls and benchmarking. | Critical for creating null models to validate low-abundance findings. |
| R/Bioconductor (with ggplot2, circlize) | Statistical computing and generation of publication-quality custom visualizations (e.g., overlap Circos plots). | Flexible environment for downstream analysis. |
| High-Performance Computing (HPC) Cluster or Cloud Instance | Provides necessary computational resources for processing large repertoire datasets and running multiple simulations. | Recommended for whole-repertoire analysis. |
| Tab-delimited Metadata File | Structured sample description file required by VDJtools to group samples for comparative analyses. | Must contain unique sample and group columns. |
Diagrams
Diagram 1: Validation Workflow for Low-Abundance Clonotypes
Diagram 2: Key VDJtools Analysis Commands
Technical Support Center
Troubleshooting Guides & FAQs
Q1: Why does my MiXCR analysis report drastically different low-abundance clonotype (<0.01%) counts when I switch from the align to the assemble function with the same dataset?
A: This discrepancy is common and stems from fundamental algorithmic differences.
align-based calling: Directly aligns reads to the reference, potentially over-inflating low-frequency clones due to PCR/sequencing errors being mis-assigned as unique clonotypes. It is less stringent at this stage.assemble-based calling: Employs a clustering and assembly step after alignment, which collapses technical variants (errors) into a consensus sequence. This aggressively filters out noise, often reducing reported low-abundance clones.
Troubleshooting Steps:
- Verify Preprocessing: Ensure identical starting FASTQ files and quality trimming parameters.
- Check Key Parameters: Compare the
--reportoutput for critical settings:-OcloneClusteringParameters(forassemble)-OqualityTrimmingParameters(for both)
- Run a Controlled Test: Process a subset with both methods and compare the top 100 clones (high abundance). If they match well, the difference is likely noise-filtering. Use the
exportClonestable for a side-by-side comparison.
Q2: How do I determine if a low-percentage clonotype (e.g., 0.001% read fraction) is a genuine rare clone or an artifact when comparing outputs from MiXCR, ImmunoSEQ, and ARResT/Interrogate?
A: Concordance across pipelines is a strong indicator of validity. Follow this diagnostic protocol:
Experimental Protocol for Cross-Pipeline Validation:
- Run each pipeline with its default, stringent settings for error correction.
- Extract the clone nucleotide sequence and its frequency from each tool's output for the clonotype in question.
- Map the sequence back to the original FASTQ files using a lightweight aligner like
bowtie2in--very-sensitive-localmode to independently verify its presence. - Check for phylogenetic support: Use a tool like
clustalto see if the low-abundance sequence has 1-2 nucleotide differences from a high-abundance clone (suggesting a hypermutation lineage).
Table 1: Low-Abundance Clone Call Comparison Across Pipelines
| Feature | MiXCR (assemble) |
ImmunoSEQ Analyzer | ARResT/Interrogate | Indicates True Clone If... |
|---|---|---|---|---|
| Default Error Correction | Yes (Clustering) | Yes (Collapsing) | Yes (Statistical) | Present in all outputs |
| Key Filtering Parameter | --min-sum-fraction |
-frequency-range |
p-value cutoff |
Consistent after relaxed filtering |
| Output Confidence Metric | Read count | Confidence score | p-value | High confidence in ≥2 pipelines |
| Best for Cross-Validation | Raw sequences | Standardized data | Advanced statistics | Sequences align to same V/J |
Q3: What is the recommended wet-lab protocol to validate bioinformatics predictions of low-frequency clones in the context of clonotypes fix research?
A: In silico findings require in vitro confirmation.
Detailed Experimental Protocol: Clone-Specific PCR & Sanger Validation
- Primer Design: Using the predicted CDR3 nucleotide sequence, design clone-specific forward primers. Use a consensus reverse primer in the constant region.
- Template Preparation: Perform nested PCR on the original cDNA sample. First round: multiplex V/J primers. Second round: clone-specific primer.
- Gel Electraction & Sequencing: Run the product on a high-percentage agarose gel, extract the band, and submit for Sanger sequencing.
- Analysis: Align the Sanger sequence to the predicted clone from the bioinformatics pipelines. A direct match confirms its physical presence.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Clonotype Validation Experiments
| Item | Function in Clonotypes Fix Research |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, Phusion) | Minimizes PCR errors during amplicon library prep for NGS, crucial for accurate low-variant calling. |
| UMI (Unique Molecular Identifier) Adapters | Tags each original mRNA molecule, allowing bioinformatics pipelines to correct for PCR and sequencing errors. |
| Clone-Specific TaqMan Probes | Enables absolute quantification via ddPCR to validate the precise frequency of a rare clone predicted computationally. |
| SPRIselect Beads | Provides reproducible size selection for NGS libraries, removing primer dimers that can complicate low-abundance analysis. |
| TRUST4 Pipeline | An independent, alignment-free software for immune repertoire analysis; used as a concordance benchmark against MiXCR. |
Visualizations
Title: Cross-Pipeline Analysis Workflow for Low-Abundance Clones
Title: Experimental Validation Protocol for Predicted Clones
Conclusion
Effectively analyzing low-percentage reads with MiXCR is not merely a technical hurdle but a gateway to discovering critical, rare immune events. By understanding the biological context, applying rigorous methodological practices, proactively troubleshooting artifacts, and validating findings against benchmarks, researchers can transform noisy low-abundance data into reliable insights. This capability is foundational for advancing precision applications in oncology (MRD), infectious disease monitoring, and immunotherapy development. Future directions will involve deeper integration of UMIs, machine learning for error suppression, and standardized reporting frameworks to ensure that the subtle but significant signals from the rare immune repertoire are consistently and accurately captured across studies.